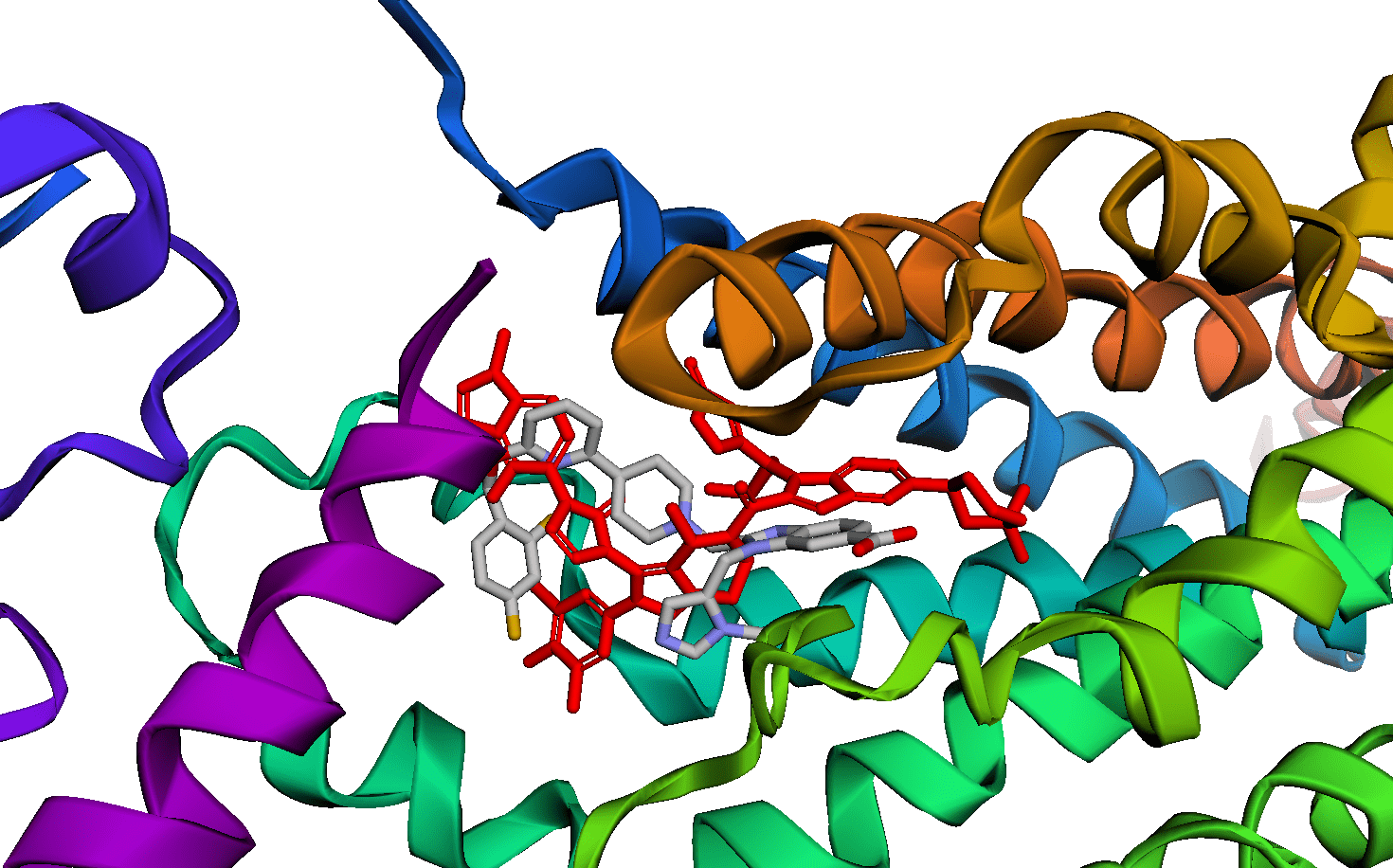
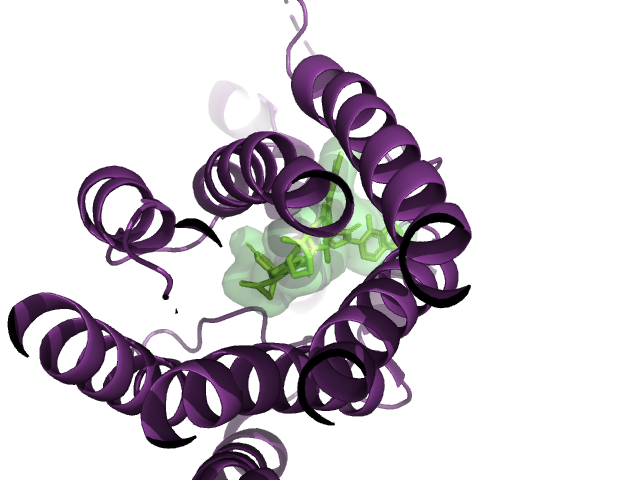
For a long time, the dream has been to be able to test code on your laptop and transparently scale it to infinite compute on the cloud. There are many, many tools that can help you do this, but modal, a new startup, comes closer to a seamless experience than anything I've used before.
There is a really nice quickstart, and they even include a generous $30/month to help get your feet wet.
pip install modal
python3 -m modal setup
git clone https://github.com/hgbrian/biomodals
cd biomodals
modal run modal_omegafold.py --input-fasta modal_in/omegafold/insulin.fasta
OmegaFold
Here's an example of how to use modal to run OmegaFold. OmegaFold is an AlphaFold/ESMFold/ColabFold-like algorithm. It is much easier to run than AlphaFold (which needs 2TB+ of reference data!) and it performs well according to a recent benchmark by 310.ai.
import glob
from subprocess import run
from pathlib import Path
from modal import Image, Mount, Stub
FORCE_BUILD = False
MODAL_IN = "./modal_in"
MODAL_OUT = "./modal_out"
stub = Stub()
image = (Image
.debian_slim()
.apt_install("git")
.pip_install("git+https://github.com/HeliXonProtein/OmegaFold.git", force_build=FORCE_BUILD)
)
@stub.function(image=image, gpu="T4", timeout=600,
mounts=[Mount.from_local_dir(MODAL_IN, remote_path="/in")])
def omegafold(input_fasta:str) -> list[tuple[str, str]]:
input_fasta = Path(input_fasta)
assert input_fasta.parent.resolve() == Path(MODAL_IN).resolve(), f"wrong input_fasta dir {input_fasta.parent}"
assert input_fasta.suffix in (".faa", ".fasta"), f"not fasta file {input_fasta}"
run(["mkdir", "-p", MODAL_OUT], check=True)
run(["omegafold", "--model", "2", f"/in/{input_fasta.name}", MODAL_OUT], check=True)
return [(pdb_file, open(pdb_file, "rb").read())
for pdb_file in glob.glob(f"{MODAL_OUT}/**/*.pdb", recursive=True)]
@stub.local_entrypoint()
def main(input_fasta):
outputs = omegafold.remote(input_fasta)
for (out_file, out_content) in outputs:
Path(out_file).parent.mkdir(parents=True, exist_ok=True)
if out_content:
with open(out_file, 'wb') as out:
out.write(out_content)
Hopefully the code is relatively self-explanatory. It's just Python code with code to set up the docker image, and some decorators to tell modal how to run the code on the cloud.
There are only really two important lines: installing OmegaFold with pip:
.pip_install("git+https://github.com/HeliXonProtein/OmegaFold.git", force_build=FORCE_BUILD)
and running OmegaFold:
run(["omegafold", "--model", "2", f"/in/{input_fasta.name}", MODAL_OUT], check=True)
The rest of the code could be left unchanged and reused for many bioinformatics tools. For example, to run minimap2 I would just add:
.run_commands("git clone https://github.com/lh3/minimap2 && cd minimap2 && make")
Finally, to run the code:
modal run modal_omegafold.py --input-fasta modal_in/omegafold/insulin.fasta
Outputs
Modal has extremely nice, almost real-time logging and billing.
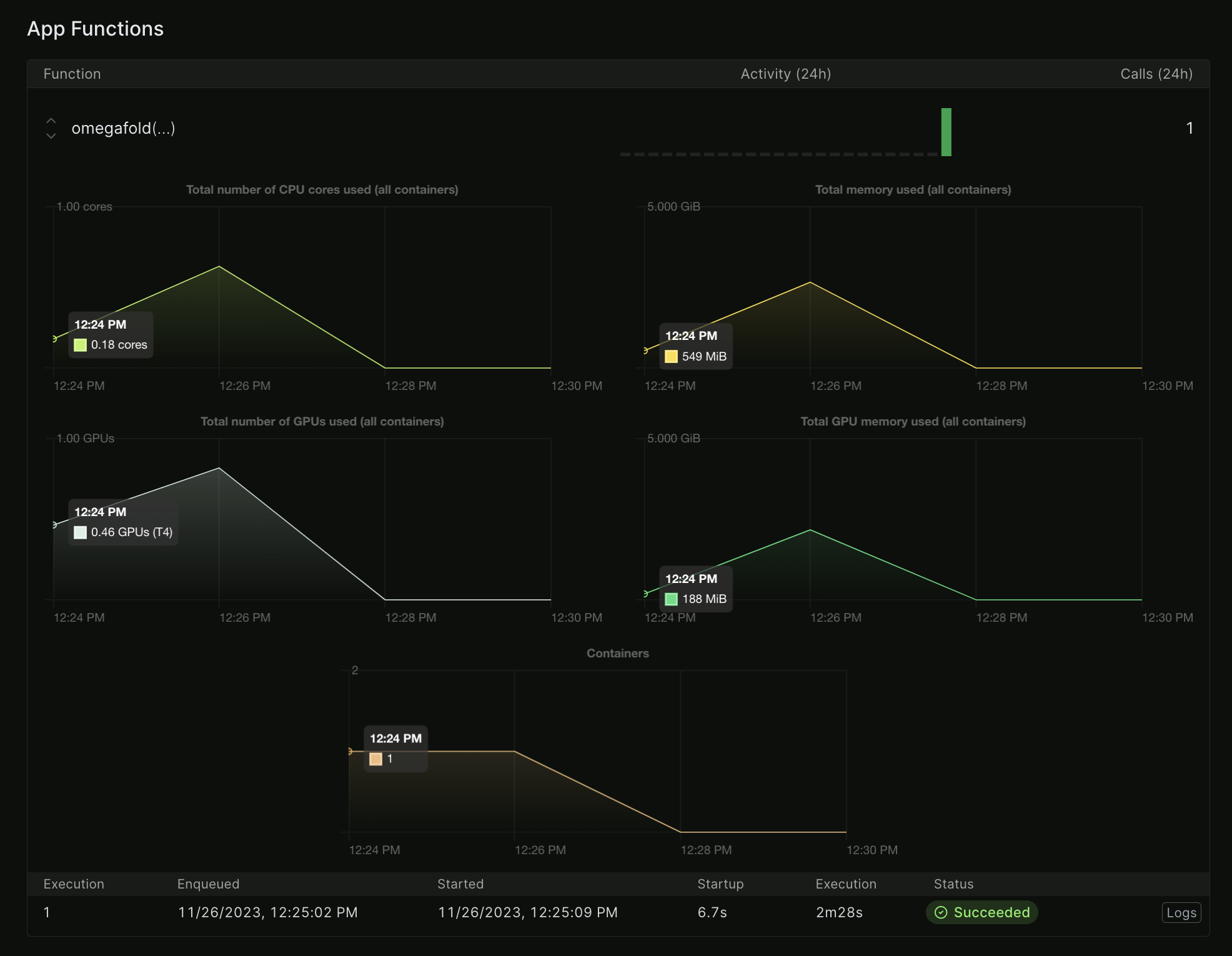
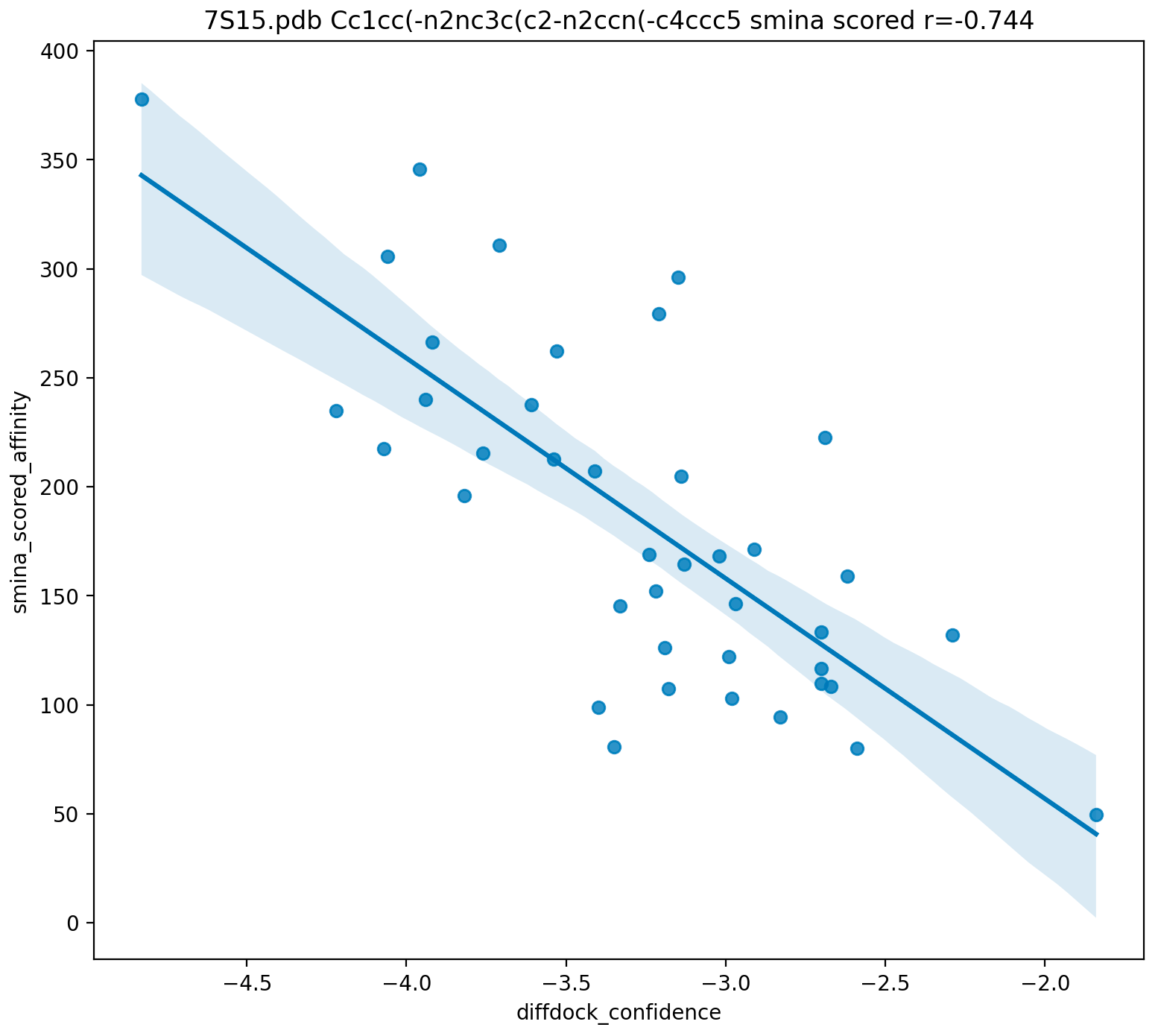
CPU and GPU usage for an OmegaFold run. Note how it tracks fractional CPU/GPU use.
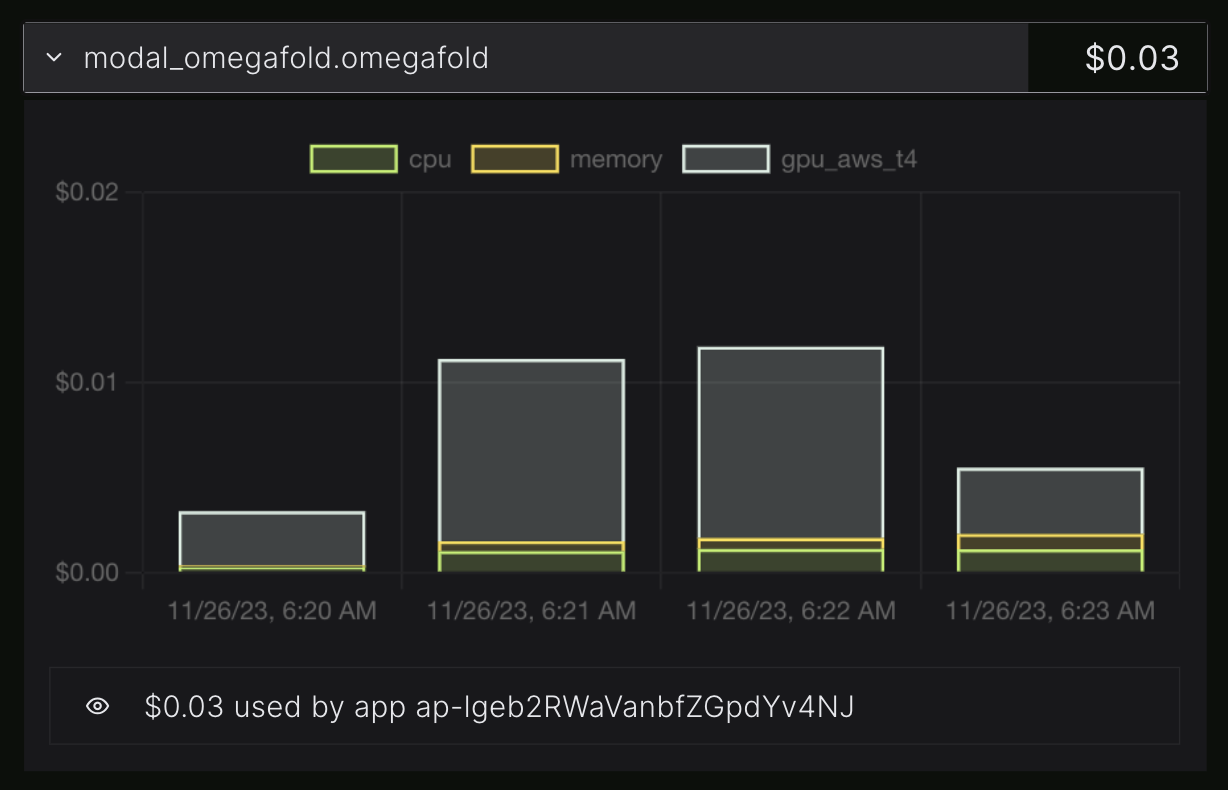
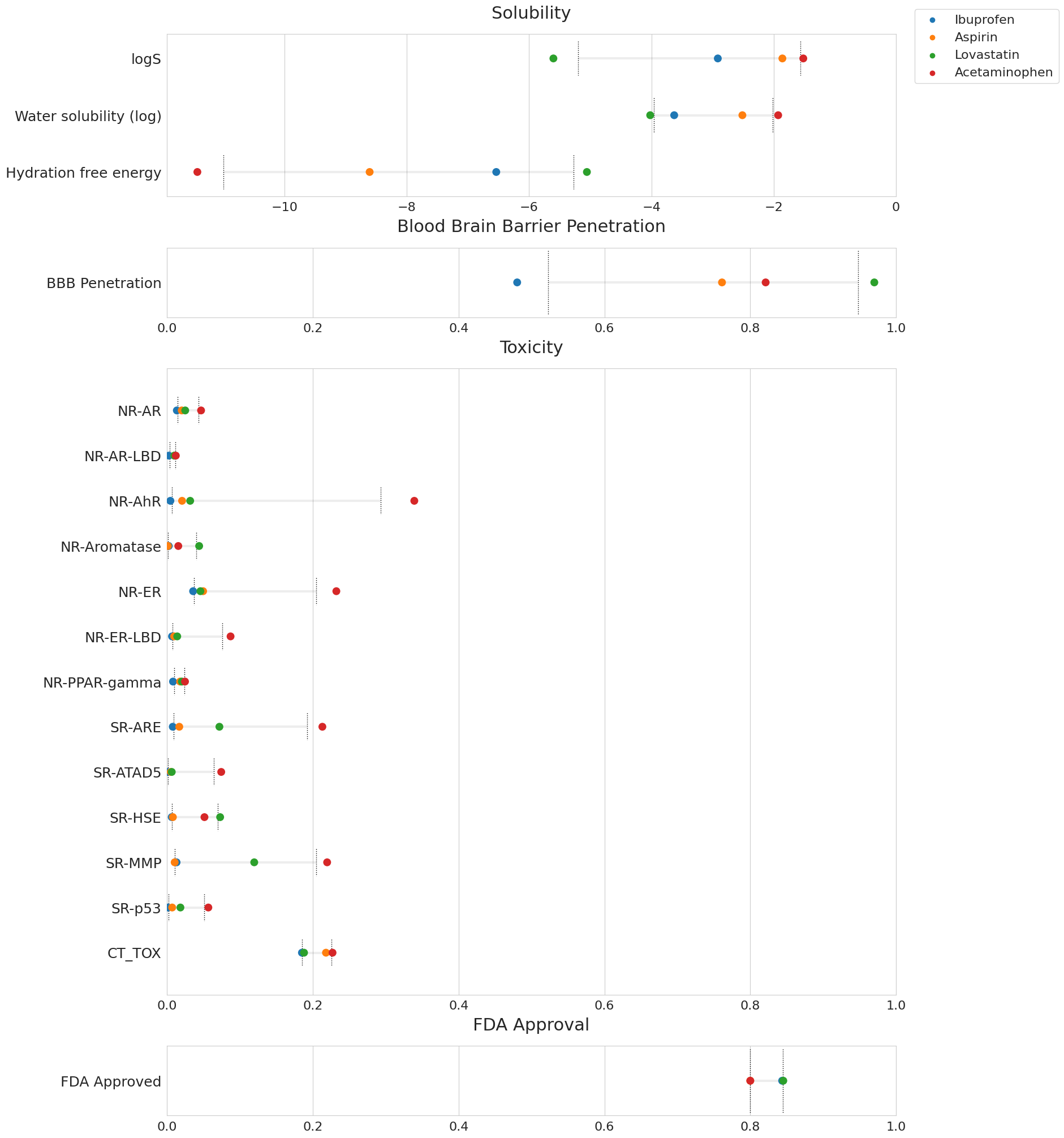
Billing information for the same run, split into CPU, GPU, RAM.
This OmegaFold run cost me 3c and took about 3 minutes. So if I wanted to run OmegaFold on a thousand proteins I could probably do the whole thing for ~$100 in minutes. (I'm not sure since my test protein is short, and I don't know how many would run in parallel.) In theory, someone reading this article could go from never having heard of modal or OmegaFold to folding thousands proteins in well under an hour. That is much faster than any alternative I can think of.
Modal vs Docker
Why not just use Docker? Docker has been around forever and you can just make a container and run it the same way! Modal even uses Docker under the hood!
There are significant differences:
- Dockerfiles are weird, have their own awful syntax, and are hard to debug;
- you have to create and manage your own images;
- you have to manage running your containers and collecting the output.
I have a direct equivalent to the above OmegaFold modal script that uses Docker, and it includes:
- a Dockerfile (30 lines);
- a Python script (80 lines, including copying files to and from buckets);
- a simple bash script to build and push the image (5 lines);
- and finally a script to run the Dockerfile on GCP (40 lines, specifying machine types, GPUs, RAM, etc).
Also, Docker can be slow to initialize, at least when I run my Docker containers on GCP. Modal has some impressive optimizations and cacheing (which I do not understand). I find I am rarely waiting more than a minute or two for scripts to start.
Modal vs snakemake, nextflow, etc
There is some overlap here, but in general the audience is different. Nextflow Tower may be a sort-of competitor, I have not tried to use it.
The advantages of these workflow systems over modal / Python scripts are mainly:
- you can separate your process into many atomic steps;
- you can parameterize different runs and keep track of parameters;
- you can cache steps and restart after a failure (a major advantage of e.g., redun).
However, for many common tasks like protein folding (OmegaFold, ColabFold), genome assembly (SPAdes), read mapping (minimap2, bwa), there is one major step — executing a binary — and it's unclear if you need to package the process in a workflow.
Pricing
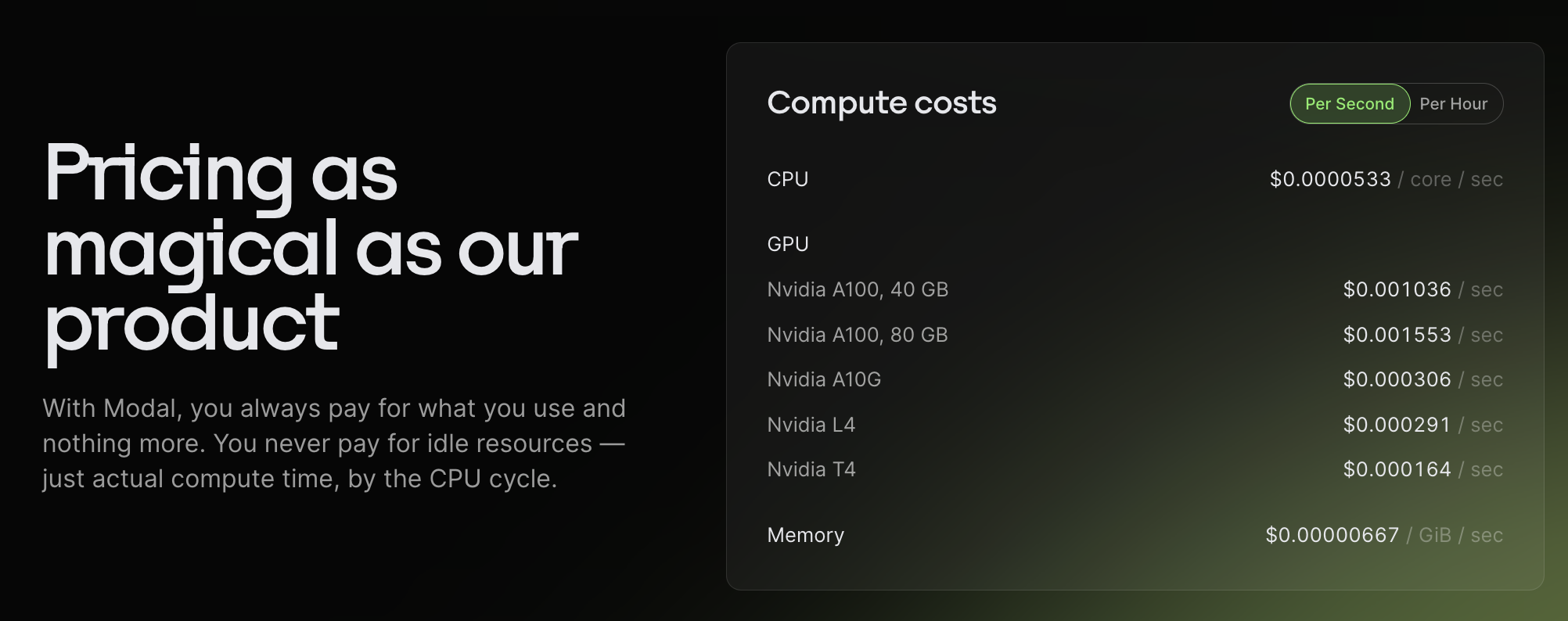
Modal is priced per second so you don't pay for more than you use.
Modal runs (transparently) on AWS, GCP, and Oracle. Another blogpost claims that Modal adds around a 12% margin. However, since Modal charges per cycle, I think it's possible you could end up saving quite a bit of money? If you run your own Docker image on a VM, you may end up paying a lot for idle time; for example, if your GPU is utilized for a fraction of the time (as with AlphaFold). It's very unclear to me how modal makes this work (or if I am misunderstanding something), but it's really nice to not have to worry about maximizing utilization.
One challenge with modal — and all cloud compute — for bioinformatics is having to push large files around (e.g., TB of sequencing reads). If you want to do that efficiently you may have to look into the modal's Volumes feature so larger files only get uploaded once.
Conclusion
Not too long ago, I felt I had to do almost everything on a linux server, since my laptop did not have enough RAM, or any kind of GPU. Now with the M-series MacBook Pros, you get a GPU and as much RAM as high end server (128GB!) I still need access to hundreds of cores and powerful GPUs, but only for defined jobs.
I am pretty excited about modal's vision of "serverless" compute. I think it's a big step forward in how to manage compute. Their many examples are illustrative. Never having to think about VMs or even having to choose a cloud is a big deal, especially these days when GPUs are so hard to find (rumor has it Oracle has spare A100's, etc!) Although it's an early startup, there is almost no lock-in with modal since everything is just decorated Python.
I made a basic biomodals repo and added some examples.