Last year I wrote a post about computational tools for drug development. Since then quite a lot has happened, especially the appearance of several generative models based on equivariant neural networks for drug design. This article is a sort of update to that post, and also a collection of colabs I have found or developed over the past year or so that can be stitched together to design drugs.
The tools listed here are focused on the earliest stages of drug development. Specifically:
- Generate a new molecule or use a virtual screen to find one
- Evaluate the molecule's potential as a drug
- Synthesize or purchase the molecule
1a. Generate a new molecule
Pocket2Mol: Protein structure → small molecules
Pocket2Mol is one of a new crop of generative models that start with a binding pocket and generate molecules that fit in the pocket. VD-Gen (with animation below) is similar to Pocket2Mol but since it has no code available so I cannot tell if it works.
I created a Pocket2Mol colab that enables easy molecule generation. The input is a PDB file and 3D coordinates to search around. The centroid of a bound ligand in the PDB file can serve as the coordinates. The output is a list of generated molecules. I rank the generated molecules with gnina, a fast, easy to install, and relatively accurate way to measure binding affinity.
ColabDesign: Protein structure → peptide binders
ColabDesign is an extremely impressive project that democratizes a lot of the recent work in generative protein models. As you may guess from the name, ColabDesign — and sister project ColabFold — allow anyone to run them via colab.

For example, even the recent state-of-the-art RFDiffusion algorithm from the Baker lab has been incorporated and made available as an RFDiffusion colab.
ColabDesign can generate proteins that conform to a specific shape or reference protein backbone (structure → sequence, i.e., AlphaFold in reverse). It can also generate peptides that can bind to a specific protein.
The same group also developed AfCycDesign, which uses a nice trick to get Alphafold to fold cyclic peptides — an increasingly important drug type, and often an alternative to antibodies. There is an AfCycDesign colab too!
In this animation it is attempting to generate a cyclic peptide covid spike protein binder, as published recently.
1b. Run a virtual screen
gnina: PDB structure + ligand → posed ligand + binding affinity
gnina is the deep-learning–based successor to the extremely popular smina, itself an AutoDock Vina fork. There is a minimal implementation available as a gnina colab. gnina is a successor to smina and appears to be strictly better. gnina can be used for virtual screening and performs very well at that.
PointVS is a new, deep-learning approach. Like many recent methods, it uses an EGNN (equivariant graph neural network). PointVS's performance is impressive, comparable to gnina, but it's unclear which method runs faster. Uni-Dock is a new GPU-accelerated Autodock Vina that claims impressive speed but unfortunately there is no open code.
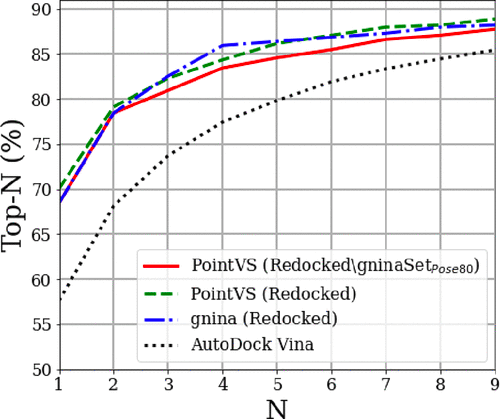
PointVS and gnina perform comparably
DiffDock: protein structure + ligand → posed ligand
DiffDock is a generative diffusion model with impressive performance. Given a protein and a ligand, DiffDock can try to find the best pose for that ligand. I created a DiffDock colab that runs DiffDock and again ranks the results using gnina.
DiffDock is a pose prediction method and is not designed to do virtual screening per se. It returns a "confidence" score that correlates with smina/gnina affinity.
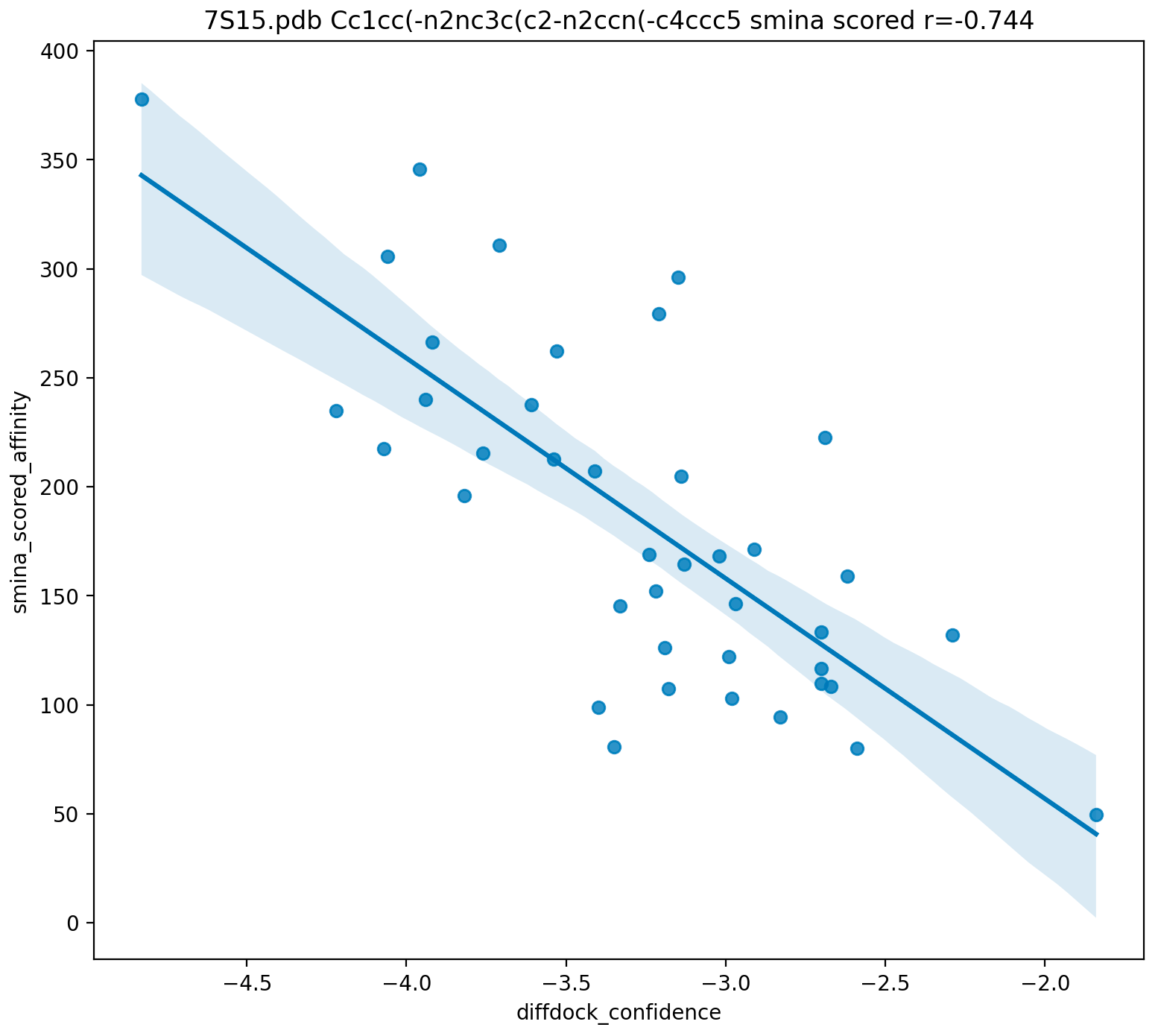
DiffDock confidence and smina/gnina binding affinities correlate fairly well
As for which algorithm produces superior results, it's unclear, but according to a recent review, gnina comes out ahead.

2. Evaluate molecule properties
SMILES property predictor: molecule → predicted properties
A molecule the binds to a protein has one property necessary to become a drug, but that is far from the only thing that matters. We can also predict a molecule's intrinsic properties using machine learning.
I created a Smiles to properties colab based on chemprop, soltrannet, and SMILES2Caption. Most of the training data is from MoleculeNet. For some reason there was no pre-trained network available so I had to retrain from scratch.
The graphs generated show the predicted properties of your SMILES as compared to FDA-approved drugs (grey bounds). In the example below, acetaminophen appears as the most toxic of the group, as expected.
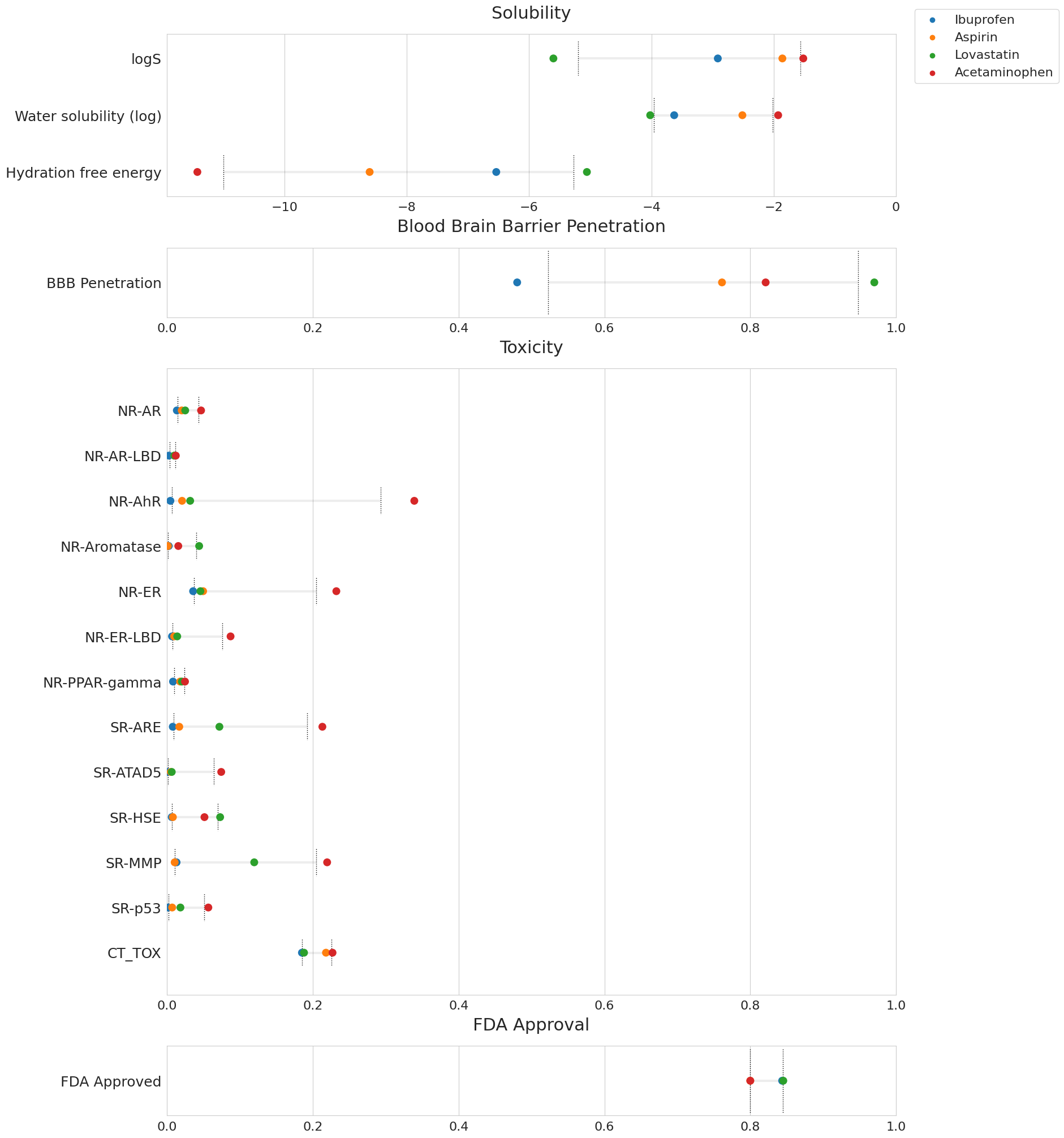
SwissADME also provides an excellent property prediction service, though anecdotally analyzing proprietary molecules on someone else's server is not commonly done in drug development.
Espaloma Charge: molecule → charge
Espaloma Charge is a standalone part of Chodera lab's Espaloma system. I include it here as they make available a very simple Espaloma Charge colab that will assign charges for your molecule.
HAC-Net: Protein structure + posed ligand → binding affinity
HAC-Net is a new deep learning method specifically designed to estimate protein–ligand binding affinity.
The HAC-Net colab takes as input a protein and posed ligand and returns a dissociation constant (pKd) and a nice pymol image. This method may or may not be more accurate than gnina — I have not attempted to benchmark.

3. Synthesize or purchase molecule
Postera Manifold: SMILES → molecule or building blocks
Postera Manifold is a machine learning tool that helps figure out how to synthesize small molecules. It can supply the building blocks and reactions necessary for synthesis, or there is a "Have PostEra make it for you" button (at least according to their documentation, but I could not find it!) However, usually synthesis of a new small molecule is a custom, CRO-driven process, that could be expensive and take a long time depending on the molecule's complexity.
Biomatik: SMILES → purchasable molecule
For peptides — which are linear, modular molecules — synthesis is much easier. A service like Biomatik (or GenScript, or many others) can supply peptides for as little as $80 per. They can use proteinogenic or non-proteinogenic amino acids, and can cyclize the peptide too.
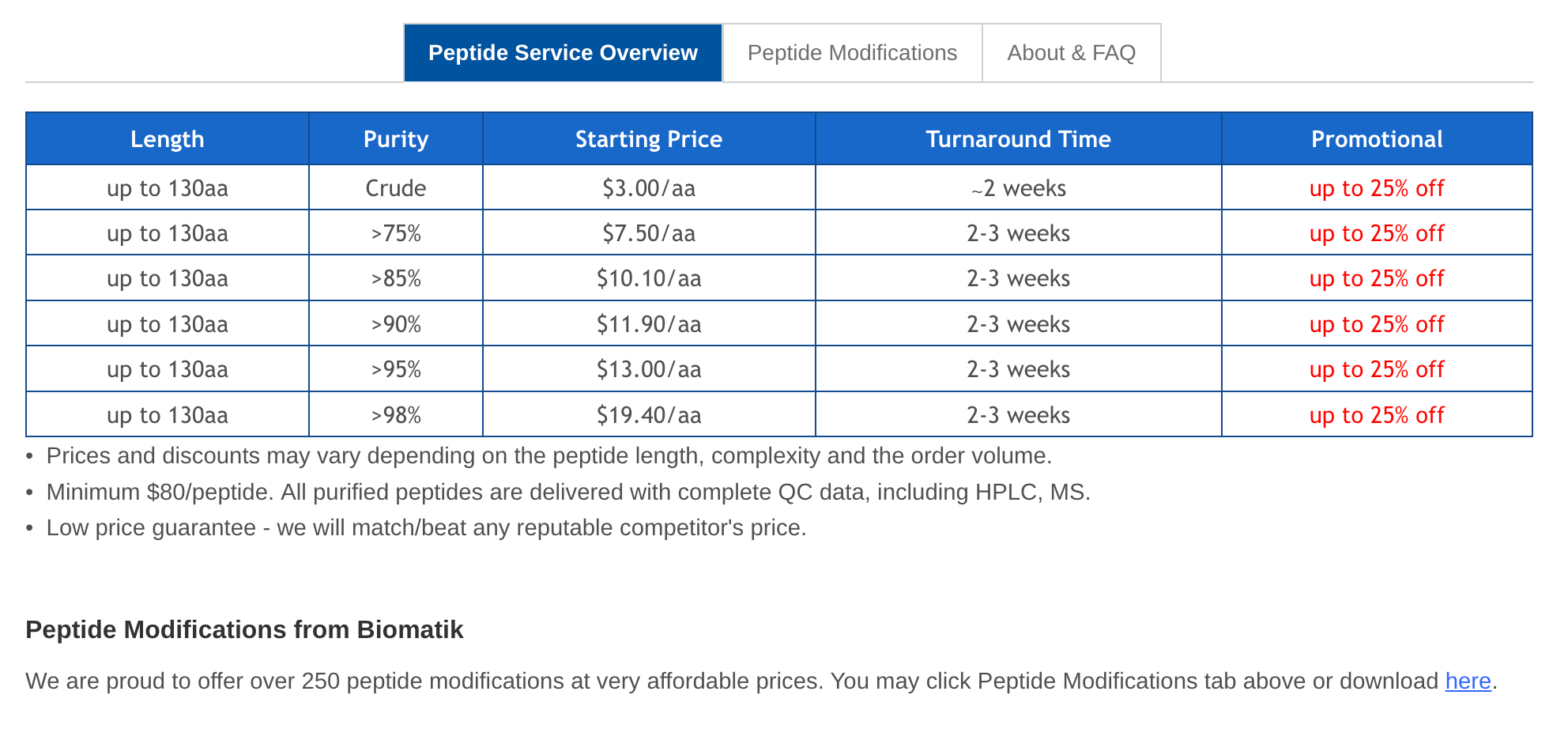
Small World: SMILES → purchasable molecule
Small world allows you to search for molecules in the largest compound databases: ZINC, Mcule, Enamine REAL... There is also a nice, unofficial Small World Python API.
If you find a close match to your designed molecule, you could evaluaate it to see if it works as a binder, or use it as a starting point for modification.
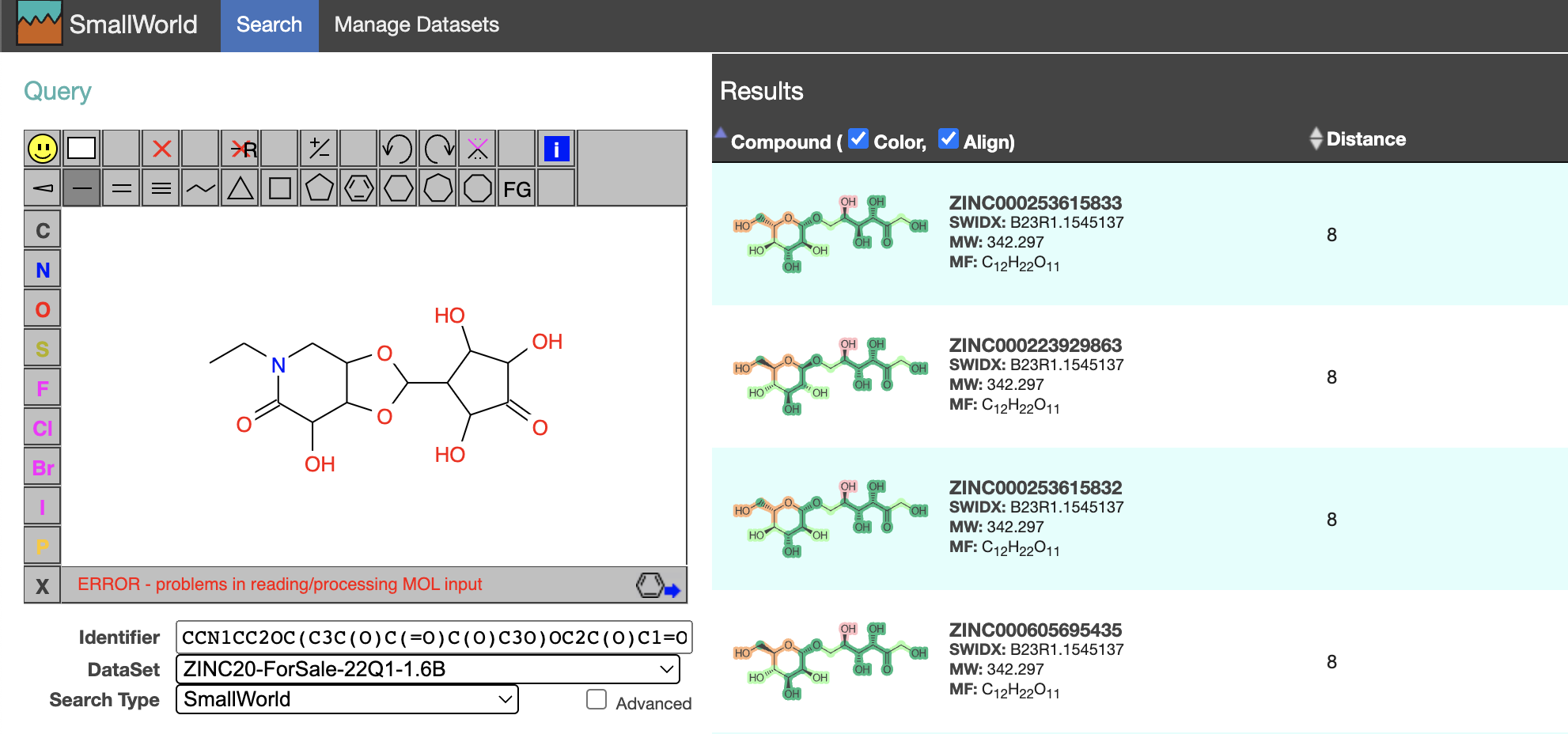
Orforglipron
As an example of using these tools, I'll use a new GLP-1 agonist, orforglipron, that showed promising results in a phase 2 study published in NEJM just this week.
First, I can generate an image of the protein GLP-1 with a different bound ligand using my pdb2png colab. (PDB:7S15, "Pfizer small molecule bound to GLP-1").
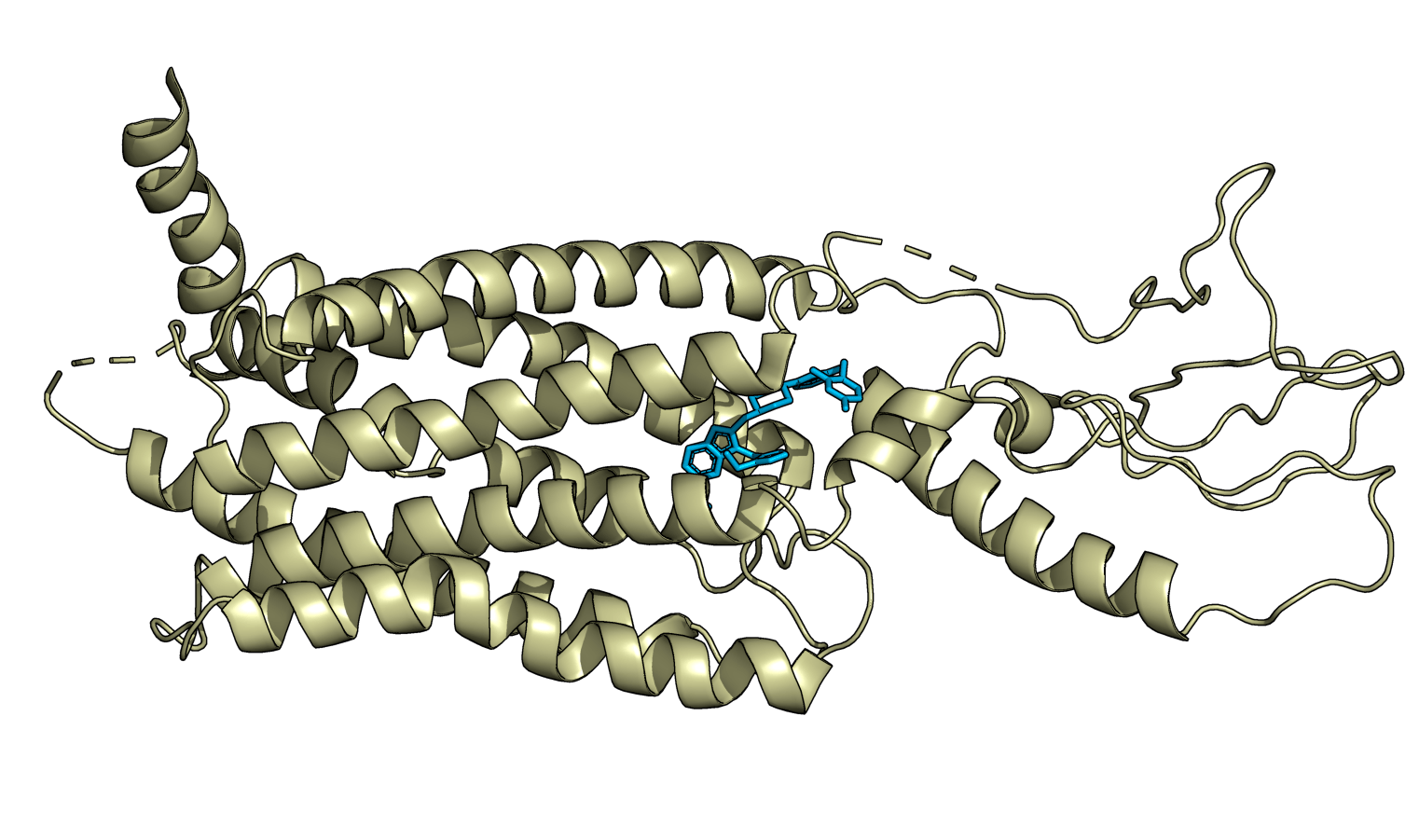
I can look at the molecule and its charges using the Espaloma Charge colab. (Nice image, but to be honest this doesn't mean much to me!)
I can get the molecule's SMILES from pubchem, and search for it using Small World.

orforglipron in Small World: dark green is a match; light green is a mismatch
A little surprisingly, I find a matching molecule that is only two edits from orforglipron in the ZINC20 "for sale" set. It is labeled as orforglipron in MCE and interestingly the text says it was "extracted from a patent". It's unclear to me why it's not an exact match. Most sources charge $2k for 1mg, but there is one source that charges $1k for 5mg.
I can evaluate its properties with SwissADME, and it's interesting to note how far outside the ideal orforglipron is, in terms of size (883 Da!) and predicted solubility.

I can use the DiffDock colab to see how well it might bind to GLP-1
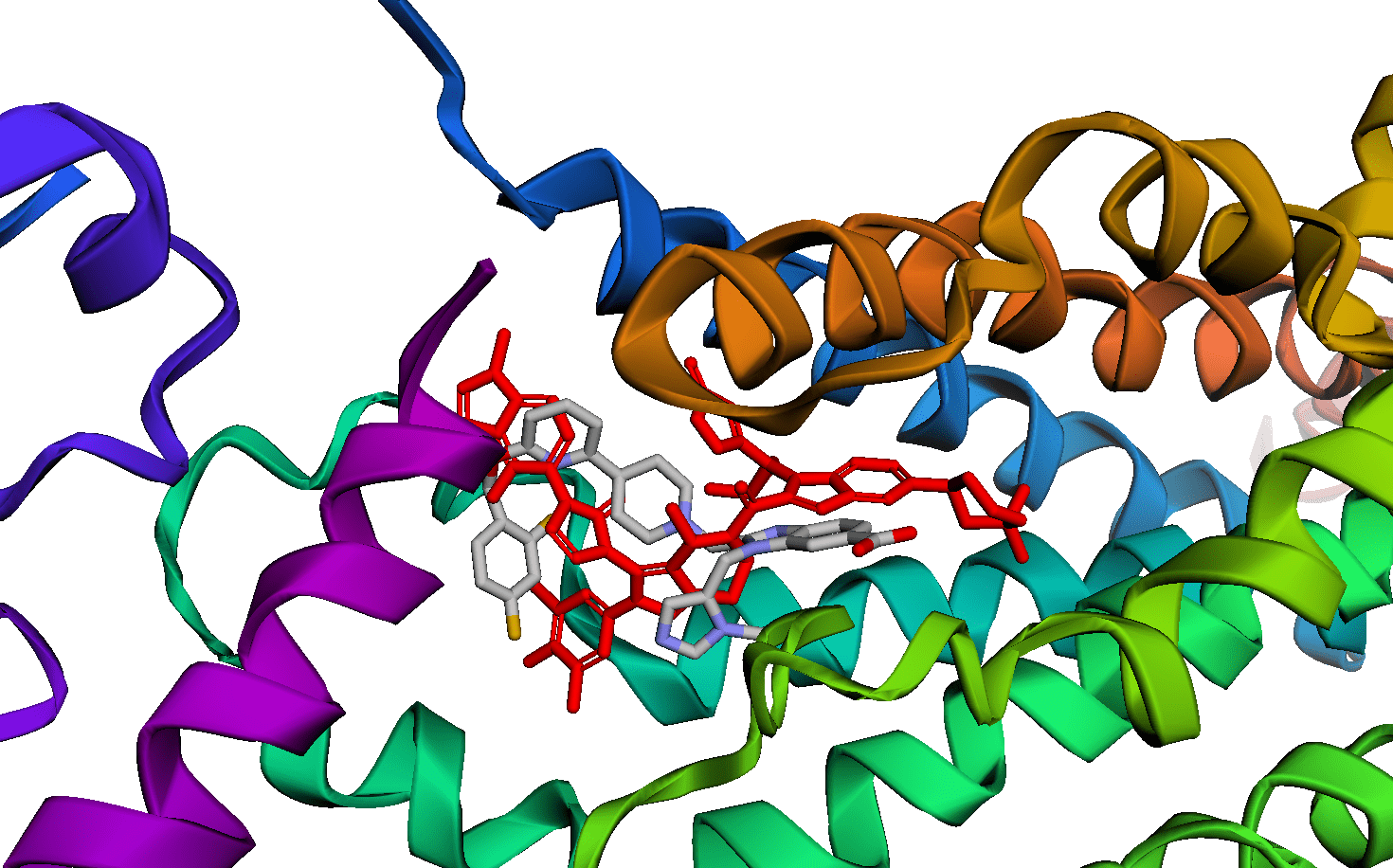
The best binding score I could get after three attempts was a DiffDock confidence of -2 (very unconfident) and a gnina affinity of -6 (poor-to-mediocre affinity). Because this molecule is so large, there are many possible conformers and it may be impossible to adequately sample them adequately. It does at least bind to the same place as the Pfizer small molecule.
When I feed this top pose from DiffDock into the HAC-Net colab, I get a pKd of 11, which is very high affinity.
The tools for drug design are getting very powerful, and colab is a fantastic way to make them widely available and easy to run. Even when I have a command-line equivalent set up, I often find running the colab to be quicker and easier. In theory, Docker provides the same kind of advantages, but it's never quite so easy, as you still have to provision compute, disk, etc. Having easy access to A100's (on the extremely affordable Colab Pro) is not bad either.