The minimal drug development pipeline is some computational tools and a full-stack CRO. I wrote about tools for virtual biotech a few years ago. Since then, quite a few new tools have emerged.
Surprisingly, since there is a pleasing analogy with cloud computing, virtual biotech has waned in popularity in recent years. It's possible that the term is just being retired, as the lines between virtual and regular biotech blur — or, it may be that a purely virtual model proves too restrictive given the laborious nature of drug development.

Atlas Venture was one of the early advocates of virtual biotech, and Atlas' Nimbus Therapeutics is one of few big successes I know of in the space. Nimbus used novel computational tools (Schrödinger's WaterMap) and CROs to develop drugs against "intractable targets". In 2016, Nimbus sold its NASH drug to Gilead for $400M upfront.
Tools
There have been exciting developments in tooling due to machine learning (especially deep learning), and the increasing importance of outsourced services in all industries.
OpenTargets: disease → protein target
Target identification has traditionally been an unstructured problem. If a target has some high-profile papers, and a clear tie to a disease, it can be blessed as a drug target. OpenTargets is an impressive effort to collate, in an unbiased way, all relevant disease–target data into one interface. On the cancer side, depmap and Project Score provide similarly impressive, and totally free, data based on CRISPR screens.

AlphaFold / OpenFold: sequence → protein structure
AlphaFold needs no introduction. It was a huge leap forward in protein structure prediction, but also a bellwether for applications of deep learning in biology.
Perhaps surprisingly, classical protein structure prediction has a mostly indirect application to drug discovery, since there are already experimental structures for most protein targets of interest (or easy-to-fold close homologs). Still, AlphaFold's influence has been immense, extending to many related areas, like antibody binding.
ProteinMPNN: protein structure → sequence
ProteinMPNN is the inverse of AlphaFold: given a structure, it can derive an amino acid sequence.
Amazingly, it only takes a few seconds to run, and the huggingface implementation includes a minimal AlphaFold implementation(!) to help validate the results. One of the proposed use-cases is to generate target-binding proteins.

OpenMM: protein structure → protein dynamics
OpenMM and HTMD are remarkably user-friendly molecular dynamics packages. With just a few lines of code, you can turn a PDB file into a full simulation of protein dynamics. MDAnalysis is a numpy-based tool for analyzing the resulting trajectories.
EquiBind: protein + ligand → docked ligand
EquiBind is a very fast, deep-learning–based ligand docking algorithm. Its performance appears to exceed the current state-of-the-art (perhaps excluding commercial tools), and because it is so fast, it's feasible to use it for virtual screening, evaluating thousands of ligands (or proteins). Docking has not had its AlphaFold moment yet, but the momentum is building, especially with developments in machine learning forcefields.

Pharmit: protein + bound ligand → alternative ligands
Pharmit, from the Koes lab, is one of my favorite drug development tools for its simple but powerful interface. You just paste in a PDB id, select a ligand of interest, and it will find similar ligands in multiple databases.
For example, searching PDB for Ivacaftor brings up this crystal structure of the drug bound to CFTR. Then pharmit allows you to find other, similar compounds that could bind at the same location.

 Ivacaftor (left) vs a similar molecule found in the molport database (right)
Ivacaftor (left) vs a similar molecule found in the molport database (right)
DeepChem / REINVENT / TorchDrug : protein structure → drug design
DeepChem, REINVENT, and TorchDrug are among the most comprehensive tools for de novo drug design. Because there are many features to optimize for a drug (logP, toxicity, etc.; see MoleculeNet for examples) these tools are really computational frameworks with many abilities.
Schrödinger, OpenEye and Acellera are more established commercial offerings in the same vein. DeepChain is a new startup providing a suite of deep learning tools, focused on protein design.
 Acellera provides some browser-based tools via playmolecule.com
Acellera provides some browser-based tools via playmolecule.com
PostEra: drug design → molecule
Designing a drug is not particularly useful unless you can synthesize and assay it. PostEra calls their service "Medicinal Chemistry powered by Machine Learning". They have a nice video explaining how it works.

 PostEra calculates a synthetic route, and enables you to have the compound synthesized.
PostEra calculates a synthetic route, and enables you to have the compound synthesized.
I have not used PostEra, but in theory, once you have designed your small molecule of interest, PostEra can synthesize it and have it delivered to your CRO. In the example above, the lead time is 6 weeks. This service could become one of the biggest enablers for virtual biotech.
CRL: molecule → assays
The easiest way to make a therapeutic is to get a CRO to do it for you. This is not as strange as it sounds. CROs employ all the kinds of people you would need to do this, and have enviable infrastructure compared to most biotechs. If you have a target, or if you have a starting point for your drug, then you could use an integrated drug discovery service for all downstream wet-lab tasks, almost completely hands-off.
Perhaps the most respected large CRO, Charles River Labs (CRL), offers such fully integrated services.
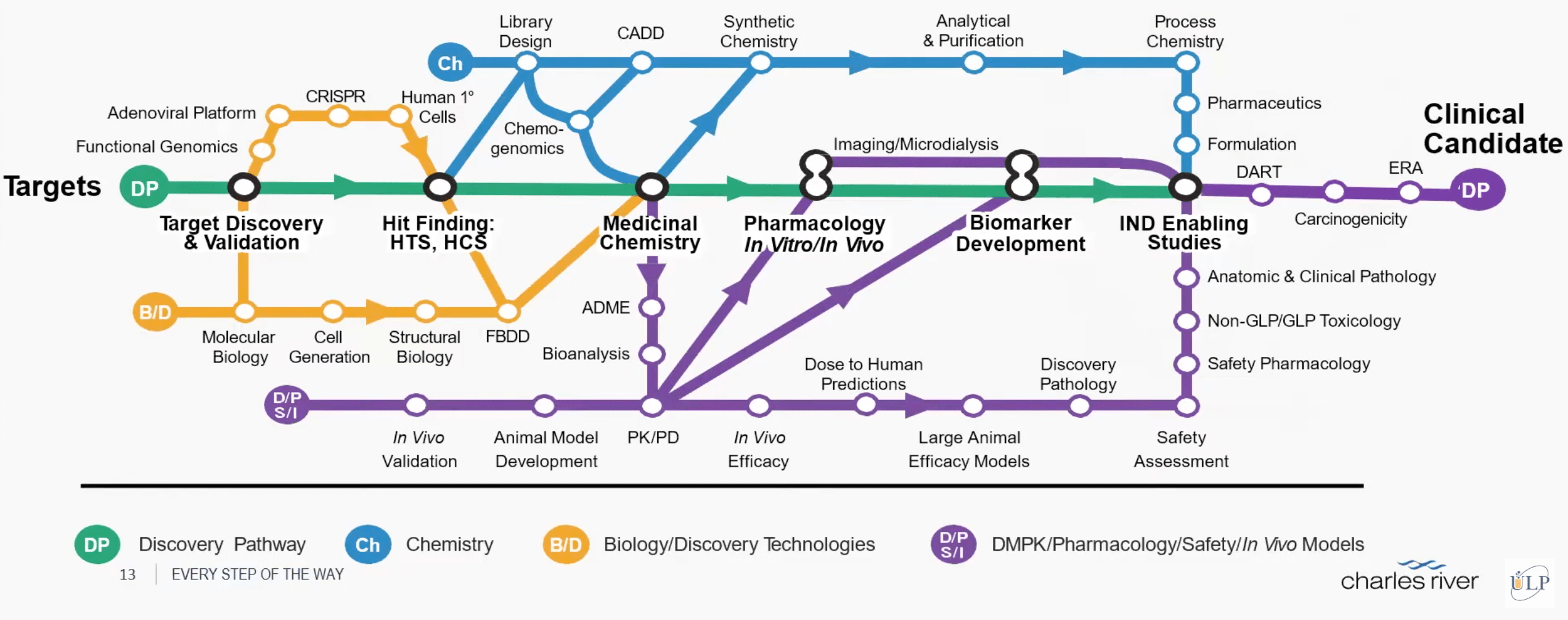
The industry standard timeline from hit identification to preclinical candidate nomination is on average 33-36 months. As a full service preclinical drug development CRO with an integrated approach to development, Charles River can reduce this timeline to as little as 24 months.
In the last 15 years Charles River has delivered 79 preclinical candidates, with over 30 of these molecules progressing to Phase I and beyond.
CRL has a range of capabilities, assembled via purchases and licensing. For example, they have deals with several cutting-edge startups, including Bit Bio for stem cell reprogramming, Atomwise for computational chemistry, and SAMDI for mass spectrometry.
Of course, you can also try to use a different CRO for each area, with the obvious benefits and drawbacks.
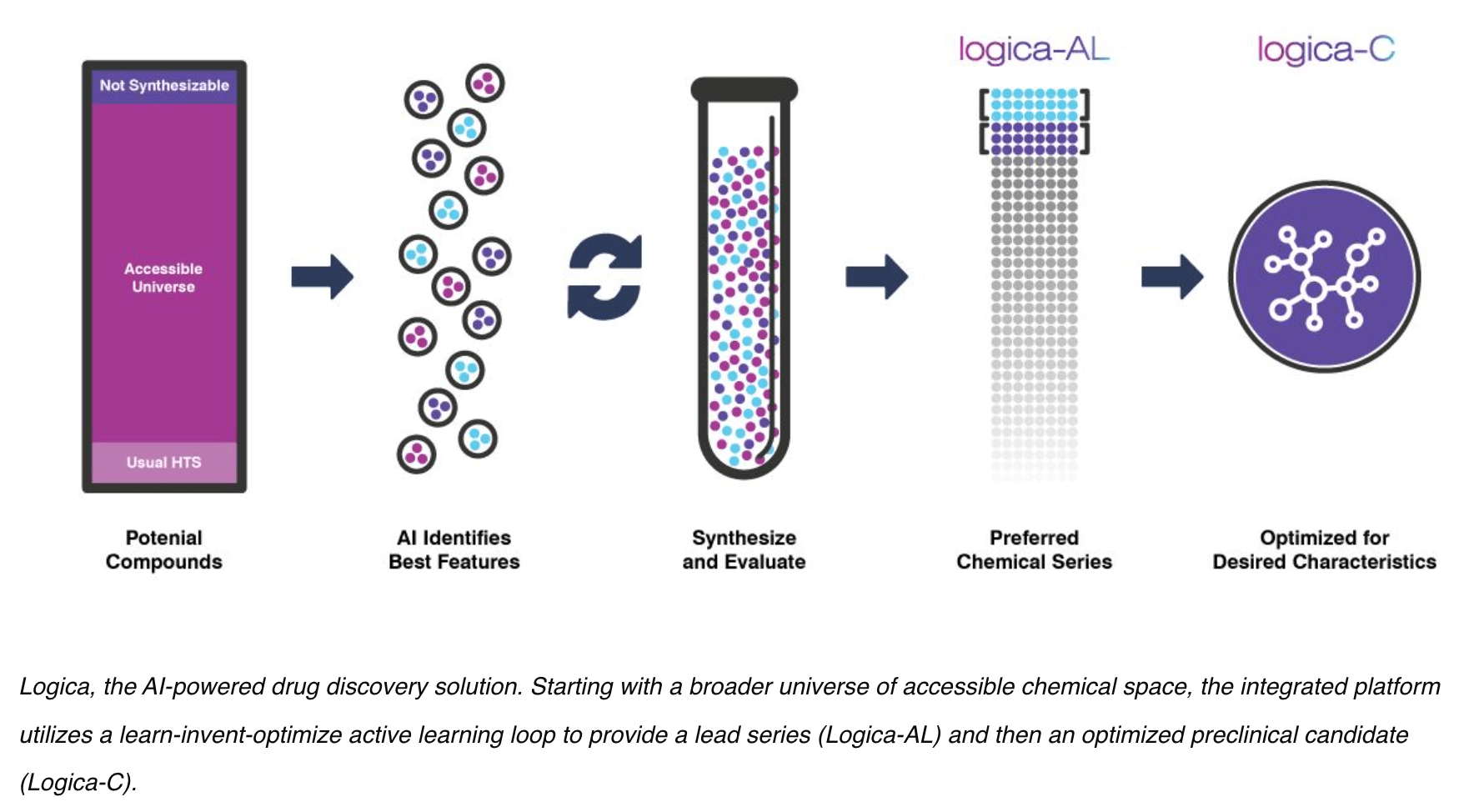
Charles River Labs offers sophisticated "AI-powered" computational chemistry services from a partnership with Valo
Joining the dots
Using the tools listed above, you could imagine the following simplified process:
- Identify a novel drug target using a database like OpenTargets.
- Derive a structure for the protein target if it does not exist.
- Examine the protein's possible conformations with OpenMM or DeepChain.
- Search for potential drugs that bind the target with Pharmit, or generate novel drugs with REINVENT. Refine the drug's molecular properties with DeepChem. Alternatively, design a short peptide that could act as a drug using ProteinMPNN and AlphaFold.
- Synthesize the compound with PostEra, or the peptide with Biomatik.
- Ship the compound directly to CRL for a specific bioassay.
- Iterate.
The success of the above scheme relies on a few things: a well-chosen drug target; computational techniques that work reasonably well; a reliable bioassay that translates well to clinical readouts. Given the right disease, target and computational approach, there is a lot of potential. This public COVID project, and the COVID moonshot (collaborating with PostEra), serve as great examples of what is possible today.