In this experiment I use standard M13 primers to amplify part of the plasmid, pUC19. In theory, these primers will amplify a ~110bp fragment.
I evaluate the experiment using a standard qPCR curve (with SYBR), and then running a gel to check the size of the amplified fragment(s).
One nice aspect of this experiment is that all of the reagents used are available in Transcriptic's standard catalog, which means the experiment can be performed remotely, without shipping anything to Transcriptic. It could be a nice test-case for a new Taq mastermix, or a new PCR protocol.
# (Python 3 setup cell omitted) # https://developers.transcriptic.com/docs/how-to-write-a-new-protocol # https://secure.transcriptic.com/_commercial/resources?q=water import json import autoprotocol from autoprotocol.protocol import Protocol p = Protocol() # 3 cols: 0=template+primers+mastermix, 1=primers+mastermix, 2=water # 3 rows: A, B, C repeated experiment_name = "puc19_m13_v1" inv = {} inv['SensiFAST SYBR No-ROX'] = "rs17knkh7526ha" inv['water'] = "rs17gmh5wafm5p" inv['M13 Forward (-20)'] = "rs17tcpupe7fdh" inv['M13 Reverse (-48)'] = "rs17tcph6e2qzh" inv['pUC19'] = "rs17tcqmncjfsh" #-------------------------------------------------------- # Provisioning things for my PCR # # Provision a 96 well PCR plate (https://developers.transcriptic.com/v1.0/docs/containers) # Type Max Dead Safe Capabilities Price # 96-pcr 160 µL 3 µL 5 µL pipette, sangerseq, spin, thermocycle, incubate, gel_separate $2.49 # pcr_plate = p.ref("pcr_plate", cont_type="96-pcr", storage="cold_4") #-------------------------------------------------------- # SYBR-including mastermix # http://www.bioline.com/us/downloads/dl/file/id/2754/sensifast_sybr_no_rox_kit_manual.pdf # Instructions: 10ul mastermix + 0.8ul primer (400nM) + 0.8ul primer (400nM) + <=8.4ul template (~100ng) + 20-vol H20 # #mastermix_tube = p.ref("mastermix_tube", cont_type="micro-2.0", storage="cold_20") for well in ["A1", "B1", "C1", "A2", "B2", "C2"]: p.provision(inv['SensiFAST SYBR No-ROX'], pcr_plate.wells(well), "10:microliter") #-------------------------------------------------------- # M13 primers # I choose m13 (-20) and (-48) because of similar Tm. This amplifies ~110bp including primers. # # 100pmol == 1ul, since Transcriptic dilutes the 1300-1900pmol into 13-19ul (depending on the primer) # I want 400nM in the final 20ul according to the SensiFAST documentation (==8pmol in 20ul) # 1ul primer in 12ul total equals 8pmol/ul # # http://www.idtdna.com/pages/products/dna-rna/readymade-products/readymade-primers # Name sequence Tm Anhyd. pmoles in 10ug # M13 Forward (-20) GTA AAA CGA CGG CCA GT 53.0 5228.5 1912.6 # M13 Forward (-41) CGC CAG GGT TTT CCC AGT CAC GAC 65.5 7289.8 1371.7 # M13 Reverse (-27) CAG GAA ACA GCT ATG AC 47.3 5212.5 1918.3 # M13 Reverse (-48) AGC GGA TAA CAA TTT CAC ACA GG 57.2 7065.7 1415.2 primer_tube = p.ref("primer_tube", cont_type="micro-2.0", storage="cold_20") p.provision(inv['M13 Forward (-20)'], primer_tube.wells(0), "1:microliter") # fwd -20 p.provision(inv['M13 Reverse (-48)'], primer_tube.wells(0), "1:microliter") # rev -48 p.provision(inv['water'], primer_tube.wells(0), "10:microliter") # water #-------------------------------------------------------- # pUC19 # 1000ug/ml -> 1ul = 1ug == 1000ng. Add 1ul to 49ul to get ~20ng/ul # Then I can transfer 5ul to get 100ng total # template_tube = p.ref("template_tube", cont_type="micro-2.0", storage="cold_20") p.provision(inv['pUC19'], template_tube.wells(0), "1:microliter") p.provision(inv['water'], template_tube.wells(0), "49:microliter") # water #-------------------------------------------------------- # Move all the reagents into the pcr plate # The "dispense" command does not work because it needs >=10ul per dispense # in increments of 5ul # for wells, ul in (["A1","B1","C1"], 4), (["A2", "B2", "C2"], 9), (["A3", "B3", "C3"], 20): for well in wells: p.provision(inv['water'], pcr_plate.wells(well), "{}:microliter".format(ul)) p.transfer(template_tube.wells(0), pcr_plate.wells(["A1", "B1", "C1"]), "5:microliter") p.transfer(primer_tube.wells(0), pcr_plate.wells(["A1", "B1", "C1", "A2", "B2", "C2"]), "1:microliter") #-------------------------------------------------------- # Thermocycle, with a hot start (95C for 2m) # Based on http://www.bioline.com/us/downloads/dl/file/id/2754/sensifast_sybr_no_rox_kit_manual.pdf # I also found http://www.environmental-microbiology.de/pdf_files/M13PCR_13jan2014.pdf # p.seal before thermocycling is enforced by transcriptic # p.seal(pcr_plate) p.thermocycle(pcr_plate, [{ "cycles": 1, "steps": [{ "temperature": "95:celsius", "duration": "2:minute"}] }, { "cycles": 40, "steps": [{ "temperature": "95:celsius", "duration": "5:second"}, { "temperature": "60:celsius", "duration": "20:second"}, { "temperature": "72:celsius", "duration": "15:second", "read": True}] }], volume="20:microliter", # volume is optional dataref="qpcr_{}".format(experiment_name), # Dyes to use for qPCR must be specified (tells transcriptic what aborbance to use?) dyes={"SYBR": ["A1", "B1", "C1", "A2", "B2", "C2", "A3", "B3", "C3"]}, # standard melting curve parameters melting_start="65:celsius", melting_end="95:celsius", melting_increment="0.5:celsius", melting_rate="5:second") #-------------------------------------------------------- # Run a gel # agarose(8,0.8%): 8 lanes, 0.8% agarose 10 minutes recommended # 10 microliters is used in the example documentation # ladder1: References at 100bp, 250bp, 500bp, 1000bp, and 2000bp. # The gel already includes SYBR green # p.gel_separate(pcr_plate.wells(["A1", "B1", "C1", "A2", "B2", "C2", "A3", "B3"]), "10:microliter", "agarose(8,0.8%)", "ladder1", "10:minute", "gel_{}".format(experiment_name)) #-------------------------------------------------------- # Analyze and output the protocol # jprotocol = json.dumps(p.as_dict(), indent=2) print(jprotocol) open("protocol.json",'w').write(jprotocol) uprint("Analyze protocol") !echo '{jprotocol}' | transcriptic analyze
Analyze protocol ---------------- ✓ Protocol analyzed 11 instructions 3 containers $25.94
new_section("Results")
After waiting several days in the Transcriptic queue for the experiment to start, the actual run took just a couple of hours.
We use 9 wells in total:
- Wells [ABC]1, "template": containing template, primers and mastermix
- Wells [ABC]2, "no template control": containing primers and mastermix
- Wells [ABC]3, "water": containing only water
Column 1: Template
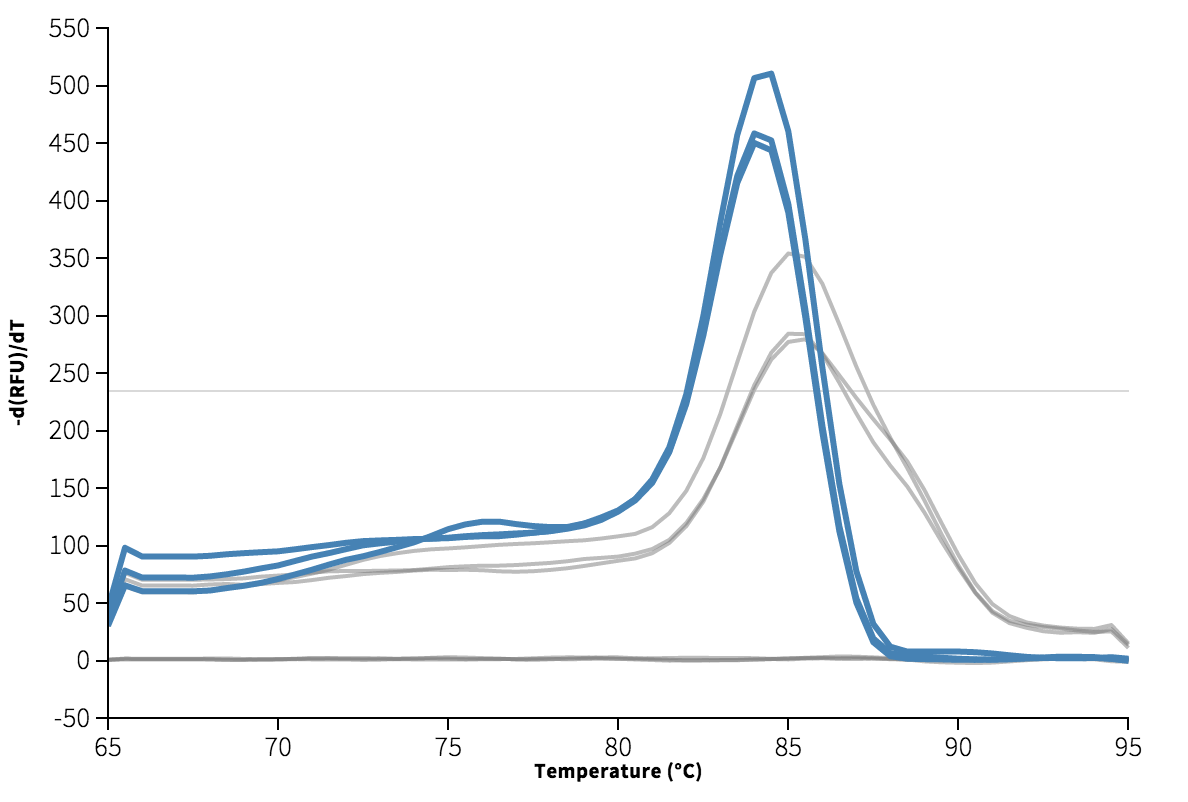
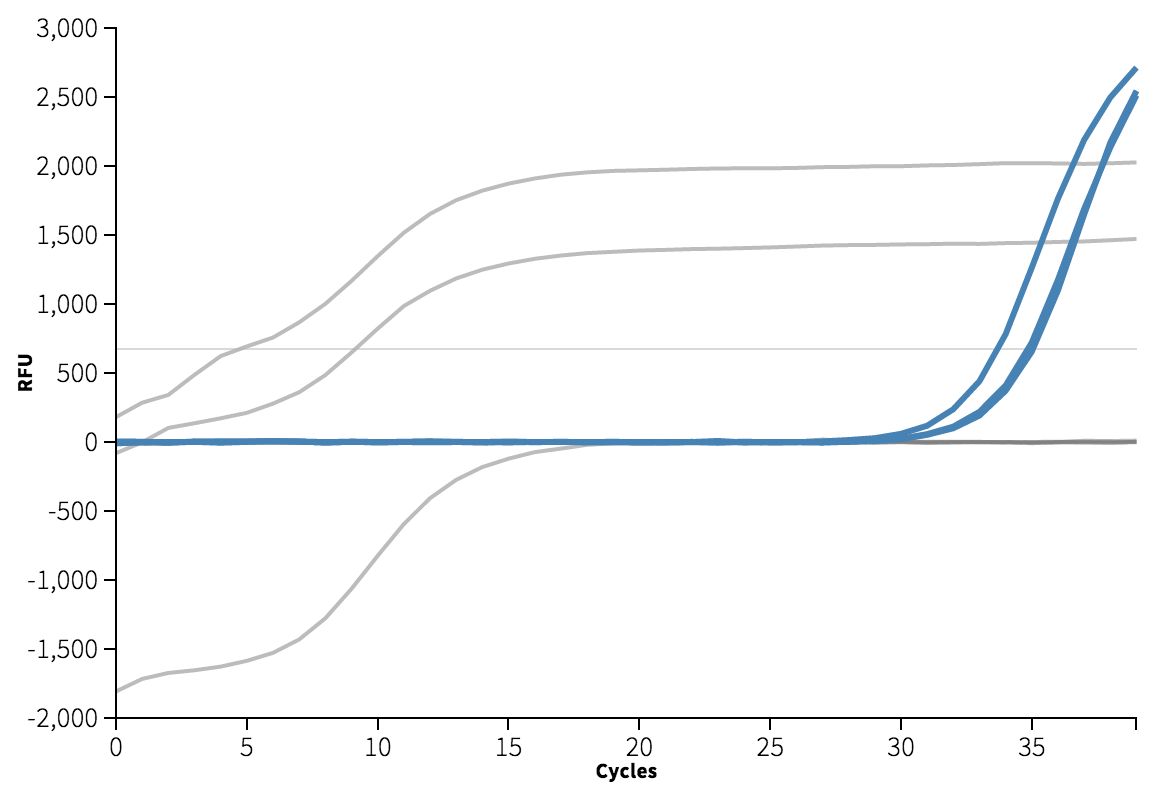
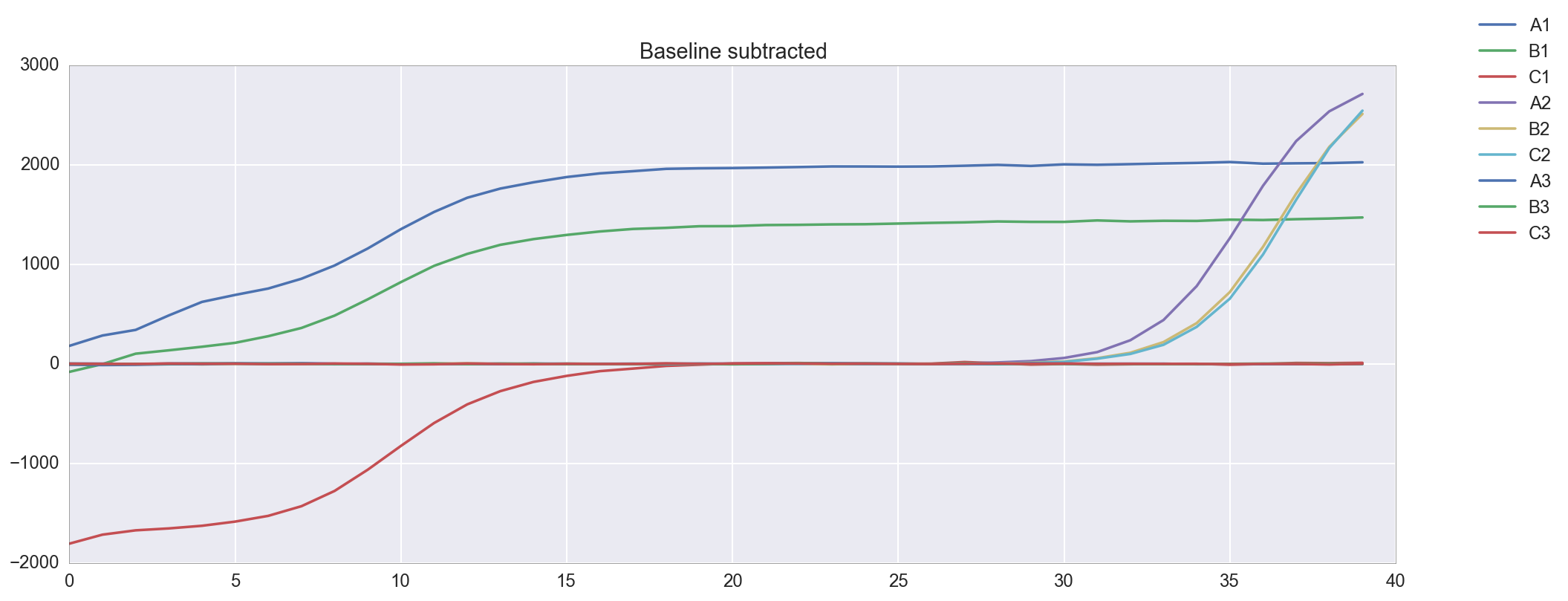
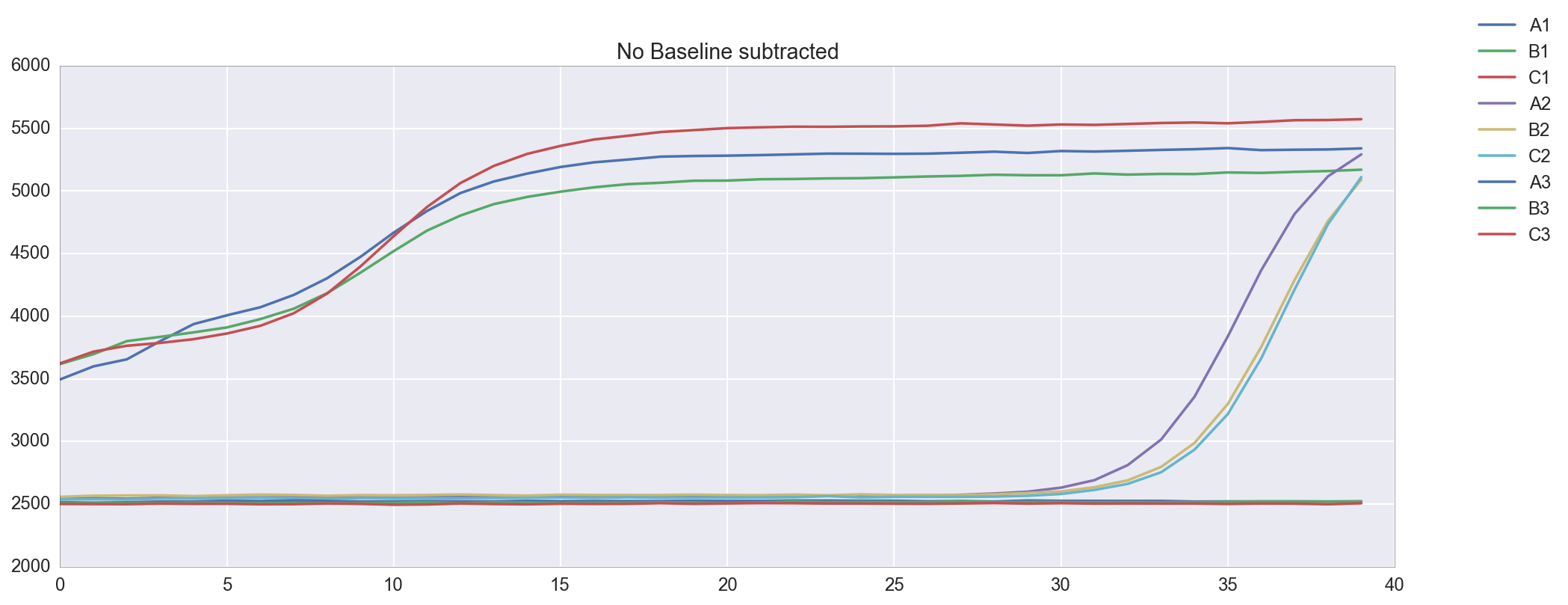
The template appears to be amplified correctly. There is an issue with the background subtraction in well C1, which has been normalized to start at about -2000 RFU. Re-graphing the raw data without background subtracted shows that this is probably a technical artifact. Unfortunately, because of this issue I do not get a Ct for well C1. The Cts are reasonably close at 4 and 9, though a <0.5 Ct difference between technical replicates is apparently desirable.
Column 2: No template control
We can see from the melting curve and amplification plots below that there is some signal in the non-template wells, indicating the presence of some double-stranded DNA. Ideally, since there is no template in these wells, we expect only single-stranded primer DNA, and no signal.
I checked the M13 primers for possible primer–dimer problems using IDT's OligoAnalyzer. If the problem were primer–dimers, we would expect the melting curve to peak at a lower temperature than the template (generally <80C), and be at least 8 Ct higher than the template. We also generally expect a deltaG below -9 kcal/mole. The lowest deltaG for these primers was far higher at -4, as you'd expect for commonly used primers.
The Ct for this amplification event is very high at 33-35, which indicates how much effort it was to amplify, and also that I could have avoided this problem by reducing the number of cycles to 30.
Column 3: Water
All of the water-only wells were blank, as expected. Although this is more of a sanity check than anything, one possible problem that could have been revealed is cross-contamination (e.g., due to reused pipette tips.)
show_html("<h1>Transcriptic plots from qPCR</h1>") show_html("<h3>No template control (A2, B2, C2) highlighted in blue</h3>") Images(["puc19_melt.png", "puc19_amp0.png"], header=["Melting curve", "Amplification"])
Transcriptic plots from qPCR
No template control (A2, B2, C2) highlighted in blue
| Melting curve | Amplification |
|---|---|
 |
 |
qPCR (Transcriptic Data API)
with open("auth.json") as auth: headers = {k:v for k,v in json.load(auth).items() if k in ["X_User_Email","X_User_Token"]} api_url = "https://secure.transcriptic.com/hgbrian/p16x6gna8f5e9/runs/r17zze6ac3b92/data.json" data_response = requests.get(api_url, headers=headers) data = data_response.json()
n_w = {str(wellnum):'ABCDEFGH'[wellnum//12]+str(1+wellnum%12) for wellnum in range(96)} w_n = {v: k for k, v in n_w.items()}
Ct = data['qpcr_puc19_m13_v1']['data']['postprocessed_data']['amp0']['SYBR']['cts'] table_print([["Well", "Ct"]] + [(n_w[k],"{:.3f}".format(v)) for k,v in sorted(Ct.items(), key=lambda x: x[1:])], header=True)
| Well | Ct |
| A1 | 4.793 |
| B1 | 9.171 |
| A2 | 33.698 |
| B2 | 34.862 |
| C2 | 35.052 |
plt.figure(figsize=(16,6)) amp0 = data['qpcr_puc19_m13_v1']['data']['postprocessed_data']['amp0']['SYBR']['baseline_subtracted'] _ = [plt.plot(amp0[w_n[well]], label=well) for well in ['A1', 'B1', 'C1', 'A2', 'B2', 'C2', 'A3', 'B3', 'C3']] _ = plt.title("Baseline subtracted") _ = plt.legend(bbox_to_anchor=(1, 1), bbox_transform=plt.gcf().transFigure) plt.figure(figsize=(16,6)) amp0 = data['qpcr_puc19_m13_v1']['data']['postprocessed_data']['amp0']['SYBR']['no_baseline_subtracted'] _ = [plt.plot(amp0[w_n[well]], label=well) for well in ['A1', 'B1', 'C1', 'A2', 'B2', 'C2', 'A3', 'B3', 'C3']] _ = plt.title("No Baseline subtracted") _ = plt.legend(bbox_to_anchor=(1, 1), bbox_transform=plt.gcf().transFigure)
Gel (Transcriptic Data API)
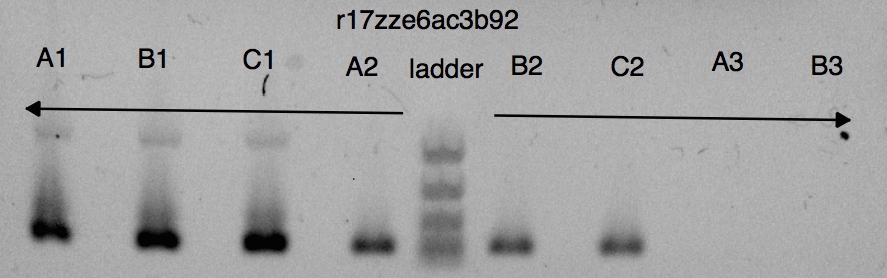
After the PCR finishes, we run a gel to see if the amplified product is the right size (approximately 100bp.)
I am guessing the gel was done manually, since there was a significant delay between the qPCR ending and the gel starting. The image also looks manually taken and is an unusual resolution. Unfortunately, it was not really possible to tell from the gel if my amplification was clean. The smallest gel ladder available on Transcriptic is 100bp-2kb, so my 100bp fragment is difficult to separate out. I can only discriminate four of the five rungs in the ladder.
The template itself can be seen at the top of the ladder in wells A1, B1, C1 (pUC19 is about 2.6kb), and the area around 100bp-250bp is darker in the wells that contain the template, so the results are reasonable.
I used SVG to rotate the image a bit, and draw horizontal lines corresponding to the rungs of the ladder.
gel_img_path = data['gel_puc19_m13_v1']['attachments'][0]['key'] with open("auth.json") as auth: headers = {k:v for k,v in json.load(auth).items() if k in ["X_User_Email","X_User_Token"]} gel_img_url = "https://secure.transcriptic.com/upload/url_for?key={}".format(gel_img_path) r = requests.get(gel_img_url, headers=headers) if r.status_code == 200: with open("gel_puc19_m13_v1.jpg", 'wb') as f: for chunk in r: f.write(chunk) Image("gel_puc19_m13_v1.jpg", width="100%")
w, h = 887, 278 # the dimensions of the image def box(xy,wh,rgba,text): box = '''<rect x="{}" y="{}" width="{}" height="{}" fill="rgba({:d},{:d},{:d},{:f})" stroke="none" /> '''.format(xy[0],xy[1], wh[0],wh[1], rgba[0],rgba[1],rgba[2],rgba[3]) text = '''<text x="{}" y="{}" text-anchor="left" font-size="12" fill="rgba({:d},{:d},{:d},{:f})"> {}</text>'''.format(xy[0]+wh[0]+10,xy[1], rgba[0], rgba[1], rgba[2], rgba[3], text) return box + text svgs = ['''<image xlink:href="static/pUC19_PCR_files/gel_puc19_m13_v1.jpg" x="0" y="0" width="{:d}px" height="{:d}px" transform="rotate(-.8)"/>'''.format(w,h)] svgs += [box((0,108), (w,1), (255,0,0,1), "start")] svgs += [box((0,156), (w,1), (0,0,255,1), "2kb")] svgs += [box((0,193), (w,1), (255,0,255,1), "1kb")] svgs += [box((0,220), (w,1), (0,255,255,1), "500bp")] svgs += [box((0,250), (w,1), (0,255,0,1), "250bp / 100bp")] show_svg(''.join(svgs), w=w+200, h=h)
new_section("Conclusions")
I was pretty happy with the results of this experiment.
Although I could not totally explain the signal in the "no template control" wells, nor competely confirm that the expected 100bp fragment was amplified from the gel image, the data was very consistent across all wells, and I believe the amplification worked fine.
This experiment cost less than $26, which is pretty good. If I could just eliminate the three day wait for the experiment to start, then this really would be cloud lab computing — the time to do the experiment is not even so different to running a typical MCMC chain!