Adaptyv is a newish startup that sells high-throughput protein assays. The major innovations are (a) they tell you the price (a big innovation for biotech services!) (b) you only have to upload protein sequences, and you get results in a couple of weeks.
A typical Adaptyv workflow might look like the following:
- Design N protein binders for a target of interest (Adaptyv has 50-100 pre-specified targets)
- Submit your binder sequences to Adaptyv
- Adaptyv synthesizes DNA, then protein, using your sequences
- In ~3 weeks you get affinity measurements for each design at a cost of $149 per data-point
This is an exciting development since it decouples "design" and "evaluation" in a way that enables computation-only startups to get one more step towards a drug (or sensor, or tool). There are plenty of steps after this one, but it's still great progress!
The Adaptyv binder design competition
A couple of months ago, Adaptyv launched a binder design competition,
where the goal was to design an EGFR binder.
There was quite a lot of excitement about the competition on Twitter,
and about 100 people ended up entering.
At around the same time,
Leash Bio launched a
small molecule competition on Kaggle,
so there was something in the air.
PAE and iPAE
For this competition, Adaptyv ranked designs based on the "PAE interaction" (iPAE) of the binder with EGFR.
PAE (Predicted Aligned Error) "indicates the expected positional error at residue x if the predicted and actual structures are aligned on residue y". iPAE is the average PAE for residues in the binder vs target. In other words, how accurate do we expect the relative positioning of binder and target to be? This is a metric that David Baker's lab seems to use quite a bit, at least for thresholding binders worth screening. It is straightforward to calculate using the PAE outputs from AlphaFold.
Unusually, compared to, say, a Kaggle competition, in this competition there are no held-out data that your model is evaluated on. Instead, if you can calculate iPAE, you know your expected position on the leaderboard before submitting.
The original paper Adaptyv reference is Improving de novo protein binder design with deep learning and the associated github repo has an implementation of iPAE that I use (and I assume the code Adaptyv use.)
Confusingly, there is also a metric called "iPAE" mentioned in the paper Systematic discovery of protein interaction interfaces using AlphaFold and experimental validation. It is different, but could actually be a more appropriate metric for binders?

At the end of last month (August 2024), there was a new Baker lab paper on Ras binders that also used iPAE, in combination with a few other metrics like pLDDT.

Experiments
A week or so after the competition ended, I found some time to try a few experiments.
Throughout these experiments, I include modal commands to run the relevant software. If you clone the biomodals repo it should just work(?)
iPAE vs Kd
The consensus seems to be that <10 represents a decent iPAE,
but in order for iPAE to be useful, it should correlate with some physical measurement.
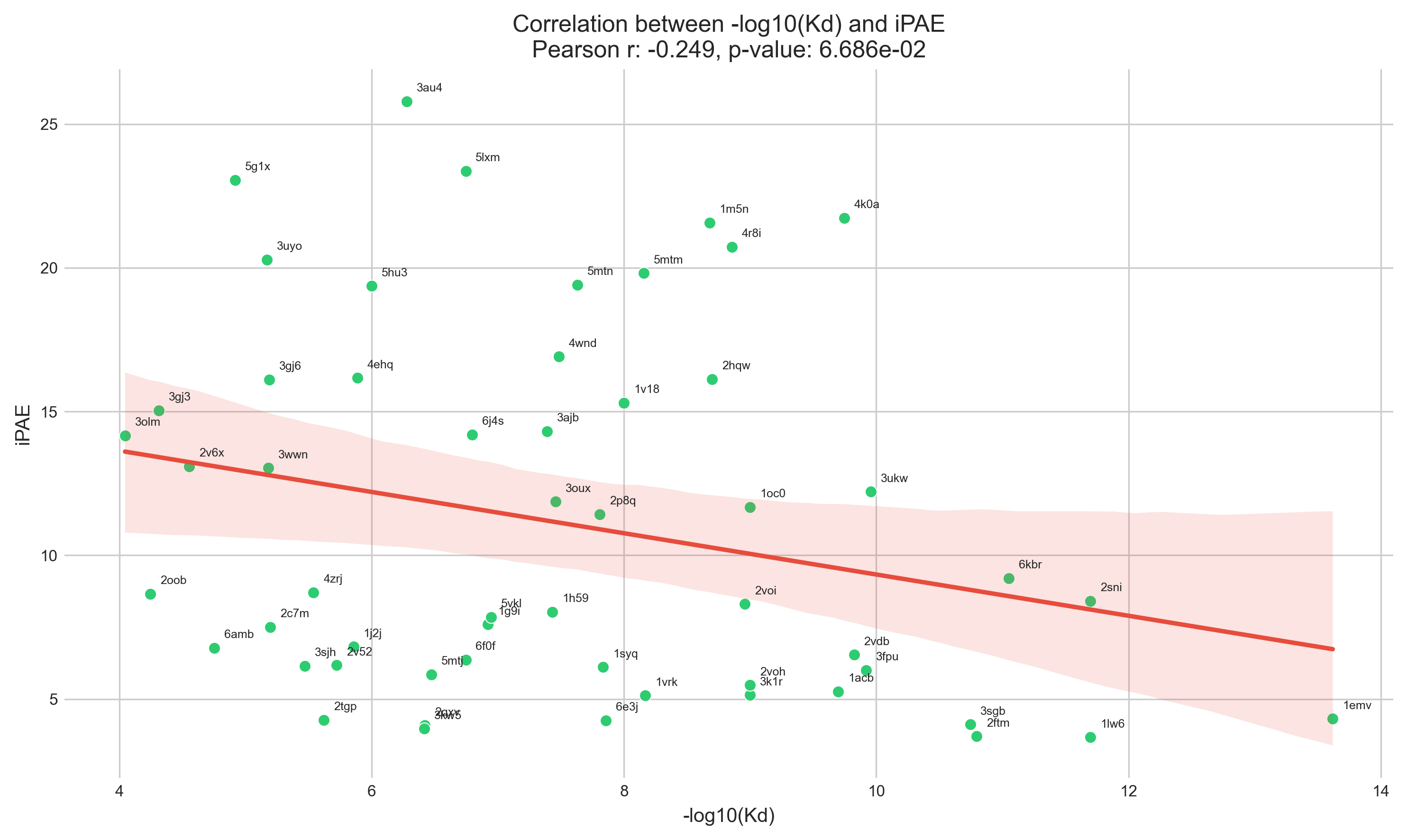
As a small experiment, I took 55 PDB entries from PDBbind
(out of ~100 binders that were <100 aas long, had an associated Kd, and only two chains),
ran AlphaFold, calculated iPAE, and correlated this to the known Kd.
I don't know that I really expected iPAE to correlate strongly with Kd,
but it's pretty weak.
PDBbind Kd vs iPAE correlation
# download the PDBbind protein-protein dataset in a more convenient format and run AlphaFold on one example
wget https://gist.githubusercontent.com/hgbrian/413dbb33bd98d75cc5ee6054a9561c54/raw -O pdbbind_pp.tsv
tail -1 pdbbind_pp.tsv
wget https://www.rcsb.org/fasta/entry/6har/display -O 6har.fasta
echo ">6HAR\nYVDYKDDDDKEFEVCSEQAETGPCRACFSRWYFDVTEGKCAPFCYGGCGGNRNNFDTEEYCMAVCGSAIPRHHHHHHAAA:IVGGYTCEENSLPYQVSLNSGSHFCGGSLISEQWVVSAAHCYKTRIQVRLGEHNIKVLEGNEQFINAAKIIRHPKYNRDTLDNDIMLIKLSSPAVINARVSTISLPTAPPAAGTECLISGWGNTLSFGADYPDELKCLDAPVLTQAECKASYPGKITNSMFCVGFLEGGKDSCQRDAGGPVVCNGQLQGVVSWGHGCAWKNRPGVYTKVYNYVDWIKDTIAANS" > 6har_m.fasta
modal run modal_alphafold.py --input-fasta 6har_m.fasta --binder-len 80
Greedy search
This is about the simplest approach possible.
- Start with EGF (53 amino acids)
- Mask every amino acid, and have ESM propose the most likely amino acid
- Fold and calculate iPAE for the top 30 options
- Take the best scoring iPAE and iterate
Each round takes around 5-10 minutes and costs around $4 on an A10G on modal.
# predict one masked position in EGF using esm2
echo ">EGF\nNSDSECPLSHDGYCL<mask>DGVCMYIEALDKYACNCVVGYIGERCQYRDLKWWELR" > esm_masked.fasta
modal run modal_esm2_predict_masked.py --input-fasta esm_masked.fasta
# run AlphaFold on the EGF/EGFR complex and calculate iPAE
echo ">EGF\nNSDSECPLSHDGYCLHDGVCMYIEALDKYACNCVVGYIGERCQYRDLKWWELR:LEEKKVCQGTSNKLTQLGTFEDHFLSLQRMFNNCEVVLGNLEITYVQRNYDLSFLKTIQEVAGYVLIALNTVERIPLENLQIIRGNMYYENSYALAVLSNYDANKTGLKELPMRNLQEILHGAVRFSNNPALCNVESIQWRDIVSSDFLSNMSMDFQNHLGSCQKCDPSCPNGSCWGAGEENCQKLTKIICAQQCSGRCRGKSPSDCCHNQCAAGCTGPRESDCLVCRKFRDEATCKDTCPPLMLYNPTTYQMDVNPEGKYSFGATCVKKCPRNYVVTDHGSCVRACGADSYEMEEDGVRKCKKCEGPCRKVCNGIGIGEFKDSLSINATNIKHFKNCTSISGDLHILPVAFRGDSFTHTPPLDPQELDILKTVKEITGFLLIQAWPENRTDLHAFENLEIIRGRTKQHGQFSLAVVSLNITSLGLRSLKEISDGDVIISGNKNLCYANTINWKKLFGTSGQKTKIISNRGENSCKATGQVCHALCSPEGCWGPEPRDCVSCRNVSRGRECVDKCNLLEGEPREFVENSECIQCHPECLPQAMNITCTGRGPDNCIQCAHYIDGPHCVKTCPAGVMGENNTLVWKYADAGHVCHLCHPNCTYGCTGPGLEGCPTNGPKIPSI" > egf_01.fasta
modal run modal_alphafold.py --input-fasta egf_01.fasta --binder-len 53
One of the stipulations of the competition is that your design must be at least 10 amino acids different to any known binder, so you must run the loop above 10 or more times. Of course, there is no guarantee that there is a single amino acid change that will improve the score, so you can easily get stuck.
After 12 iteratations (at a cost of around $50 in Alphafold compute), the best score I got was 7.89, which would have been good enough to make the top 5. (I can't be sure, but I think my iPAE calculation is identical!) Still, this is really just brute-forcing EGF tweaks. I think the score was asymptoting, but there were also jumps in iPAE with certain substitutions, so who knows?
Unfortunately, though the spirit of the competition was to find novel binders, the way iPAE works means that the best scores are very likely to come from EGF-like sequences (or other sequences in AlphaFold's training set).
Adaptyv are attempting to mitigate this issue by (a) testing the top 200 and (b) taking the design process into account. It is a bit of an impossible situation, since the true wet lab evaluation happens only after the ranking step.
Bayesian optimization
Given an expensive black box like AlphaFold + iPAE, some samples, and a desire to find better samples, one appropriate method is Bayesian optimization.
Basically, this method allows you, in a principled way, to control how much "exploration" of new space is appropriate (looking for global minima) vs "exploitation" of variations on the current best solutions (optimizing local minima).
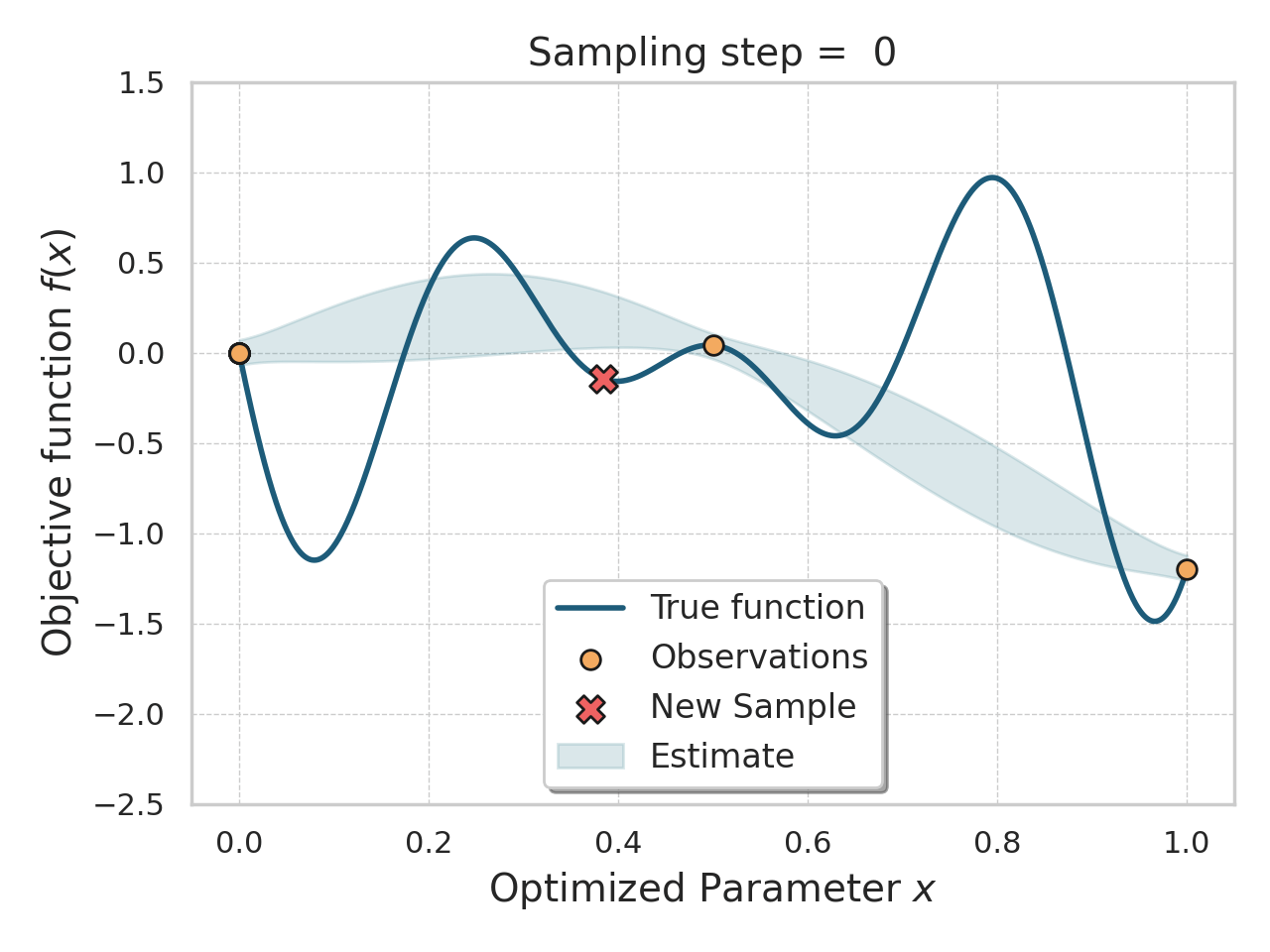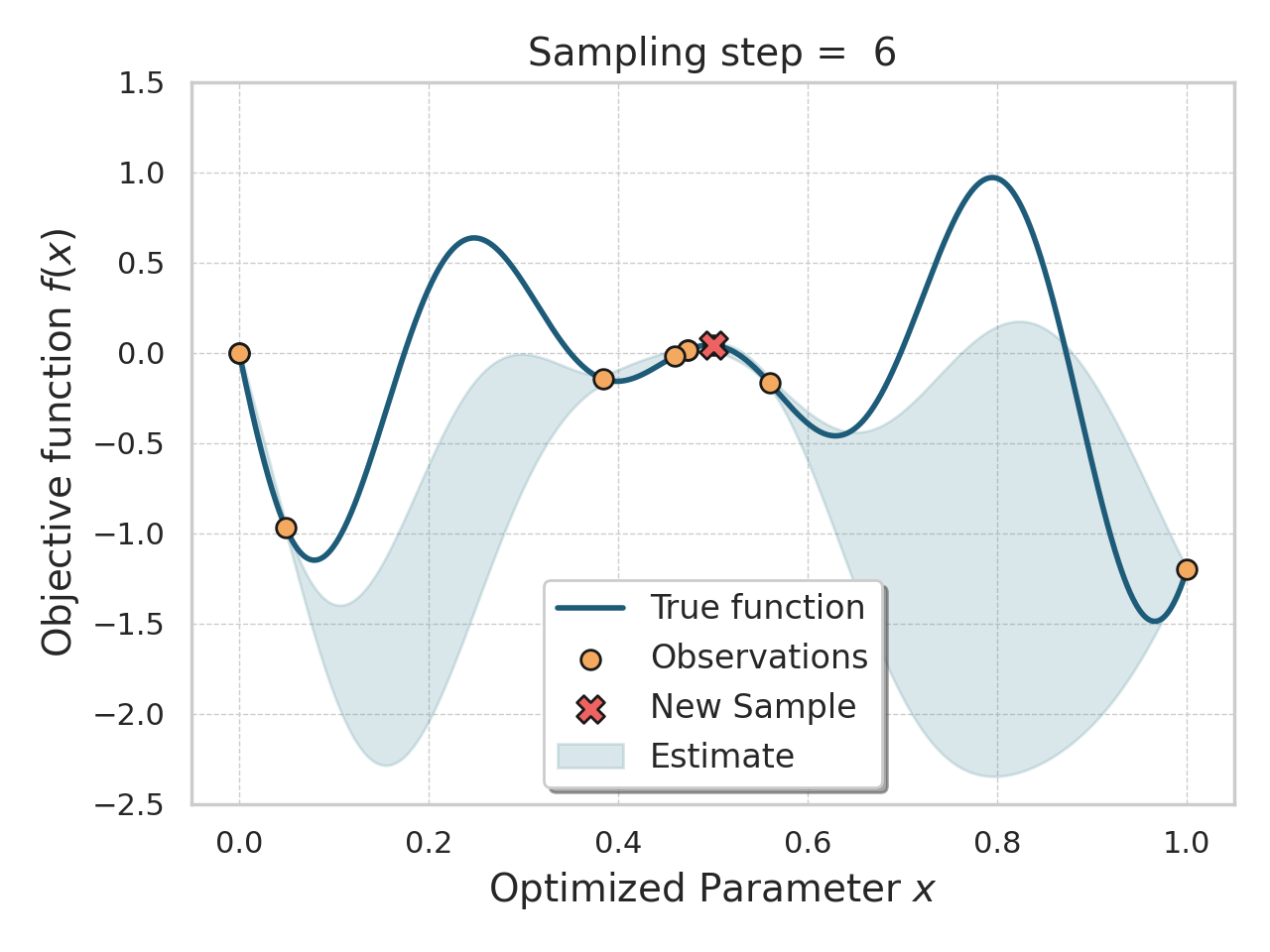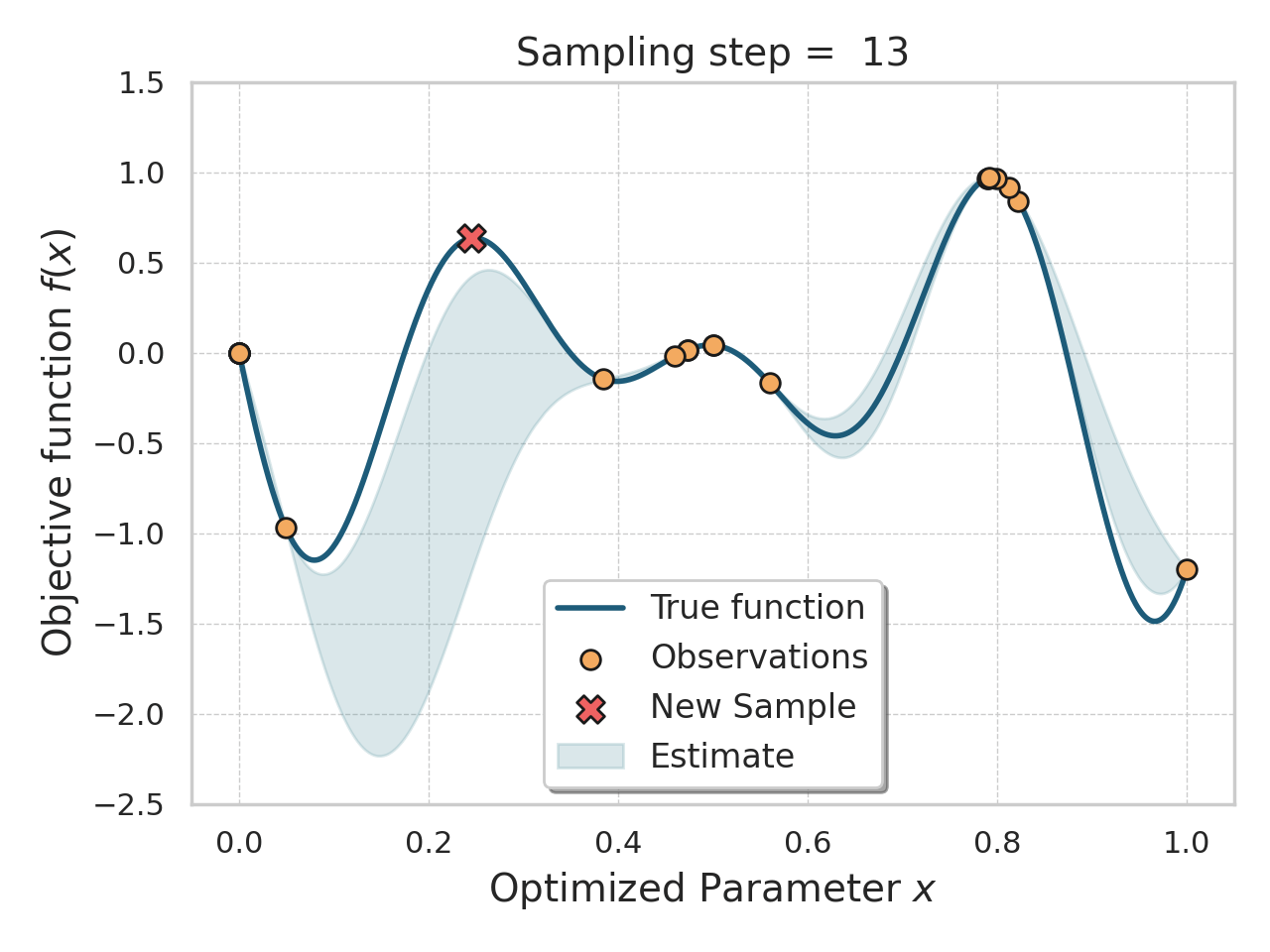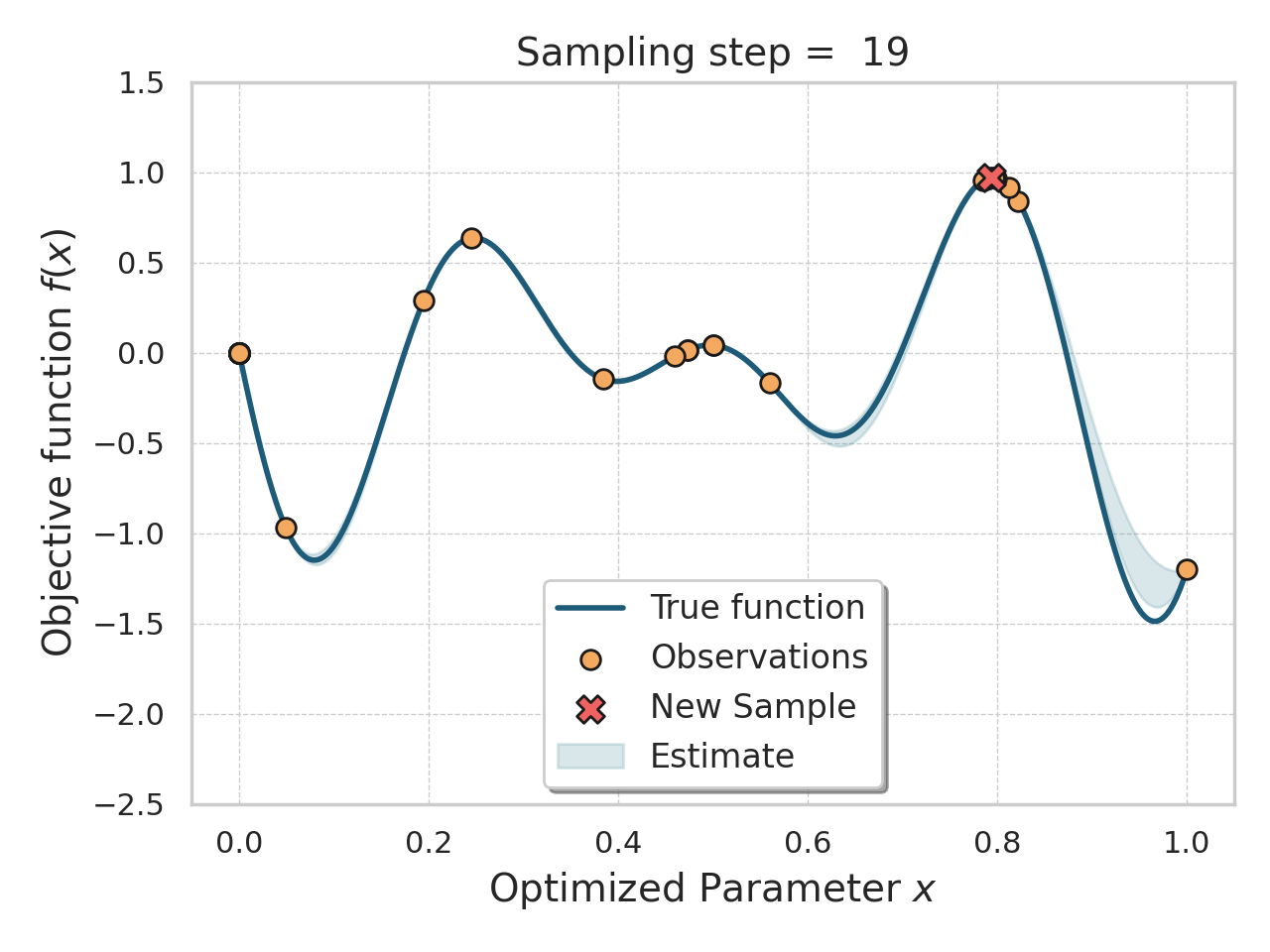
Bayesian optimization of a 1D function
The input to a Bayesian optimization is of course not amino acids, but numbers, so I thought reusing the ESM embeddings would be a decent, or at least convenient, idea here.
I tried both the Bayesian Optimization package and a numpyro Thompson sampling implementation. I saw some decent results at first (i.e., the first suggestions seemed reasonable and scored well), but I got stuck either proposing the same sequences over and over, or proposing sequences so diverged that testing them would be a waste of time. The total search space is gigantic, so testing random sequences will not help. I think probably the ESM embeddings were not helping me here, since there were a lot of near-zeros in there.
This is an interesting approach, and not too difficult to get started with, but I think it would work better with much deeper sampling of a smaller number of amino acids, or perhaps a cruder, less expensive, evaluation function.
ProteinMPNN
ProteinMPNN (now part of the LigandMPNN package), maps structure to sequence (i.e., the inverse of AlphaFold). For example, you can input an EGF PDB file, and it will return a sequence that should produce the same fold.
I found that for this task ProteinMPNN generally produced sequences with low confidence (as reported by ProteinMPNN), and as you'd expect, these resulted in low iPAEs. Some folds are difficult for ProteinMPNN, and I think EGF falls into this category. To run ProteinMPNN, I would recommend Simon Duerr's huggingface space, since it has a friendly interface and includes an AlphaFold validation step.
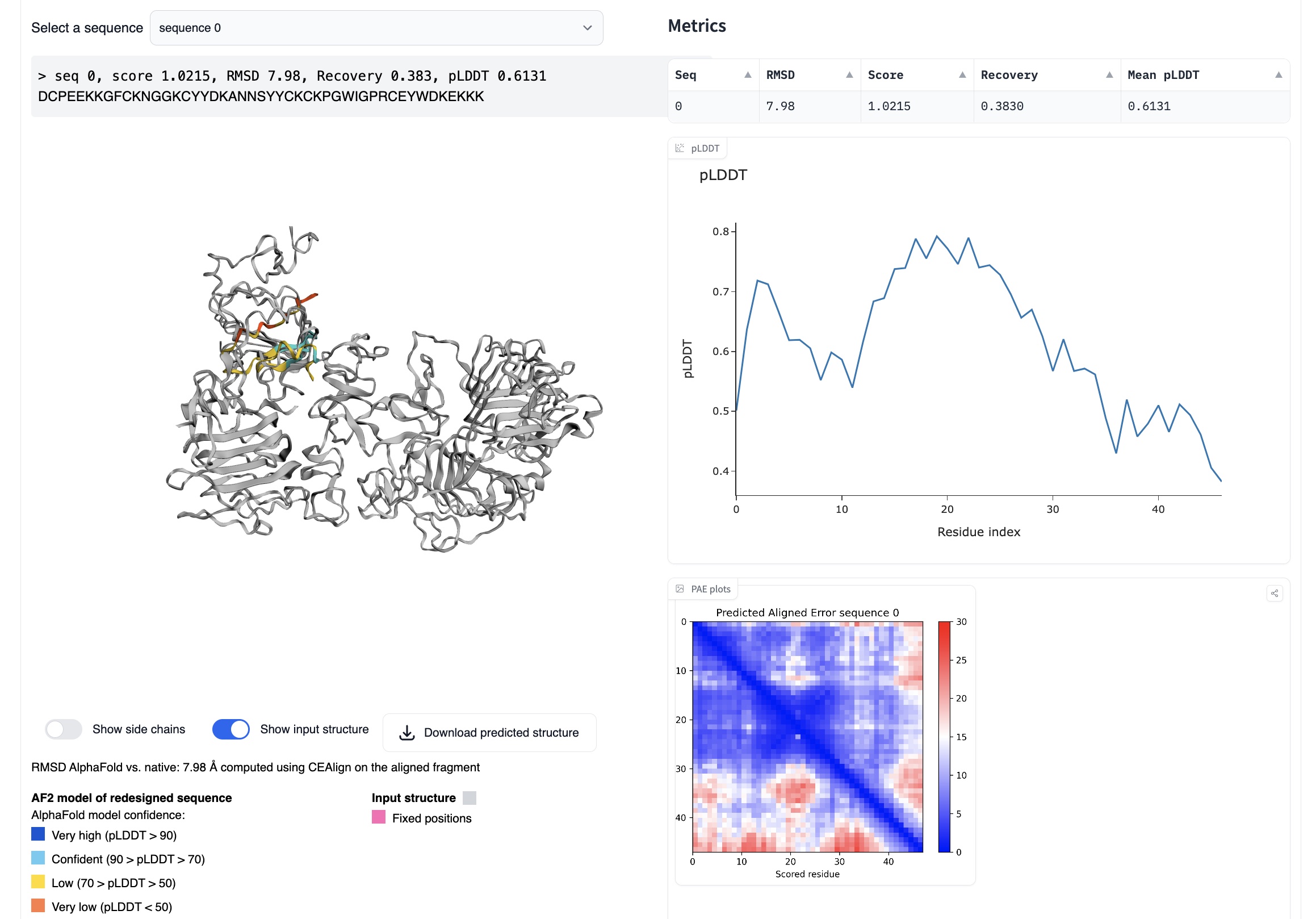
ProteinMPNN running on huggingface
# download a EGF/EGFR crytal structure and try to infer a new sequence that folds to chain C (EGF)
wget https://files.rcsb.org/download/1IVO.pdb
modal run modal_ligandmpnn.py --input-pdb 1IVO.pdb --extract-chains AC --params-str '--seed 1 --checkpoint_protein_mpnn "/LigandMPNN/model_params/proteinmpnn_v_48_020.pt" --chains_to_design "C" --save_stats 1 --batch_size 5 --number_of_batches 100'
RFdiffusion
RFdiffusion was the first protein diffusion method that showed really compelling results in generating de novo binders. I would recommend ColabDesign as a convenient interface to this and other protein design tools.
The input to RFdiffusion can be a protein fold to copy, or a target protein to bind to, and the output is a PDB file with the correct backbone co-ordinates, but with every amino acid labeled as Glycine. To turn this output into a sequence, this PDB file must then be fed into ProteinMPNN or similar. Finally, that ProteinMPNN output is typically folded with AlphaFold to see if the fold matches.
Although RFdiffusion massively enriches for binders over random peptides, you still have to screen many samples to find the really strong binders. So, it's probably optimistic to think that a few RFdiffusion-derived binders will show strong binding, even if you can somehow get a decent iPAE.

In my brief tests with RFdiffusion here, I could not generate anything that looked reasonable. I think in practice, the process of using RFdiffusion successfully is quite a bit more elaborate and heuristic-driven than anything I was going to attempt.
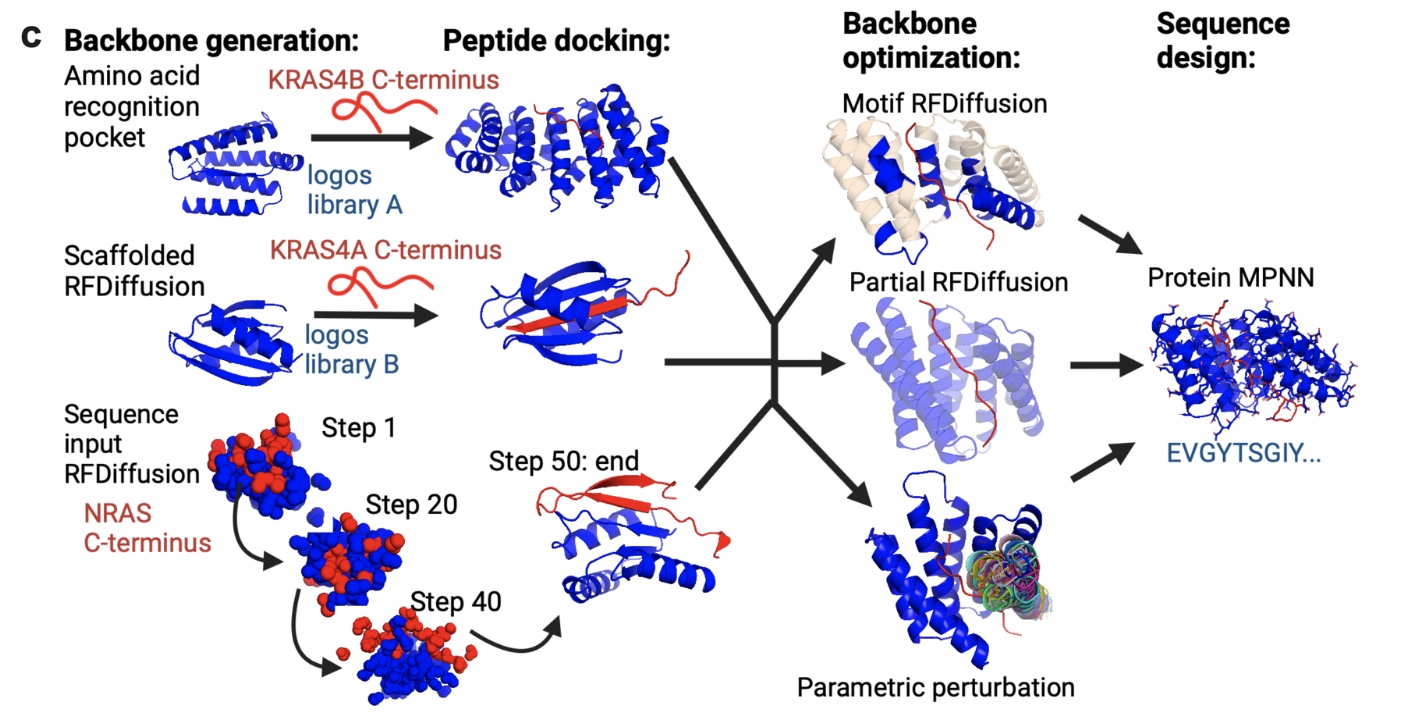
Figure 1 from De novo design of Ras isoform selective binders, showing multiple methods for running RFdiffusion
# Run RFdiffusion on the EGF/EGFR crystal structure, and diffuse a 50-mer binder against chain A (EGFR)
modal run modal_rfdiffusion.py --contigs='A:50' --pdb="1IVO"
Other things
A few other strategies I thought might be interesting:
- Search FoldSeek for folds similar to EGF. The idea here is that you might find a protein in another organism that wants to bind EGFR. I do find some interesting human-parasitic nematode proteins in here, but decided these were unlikely to be EGFR binders.
- Search NCBI for EGF-like sequences with blastp. You can find mouse, rat, chimp, etc. but nothing too interesting. The iPAEs are lower than human EGF, as expected.
- Search the patent literature for EGFR binders. I did find some antibody-based binders, but as expected for folds that AlphaFold cannot solve, the iPAE was low.
- Delete regions of the protein with low iPAE contributions to increase the average score. I really thought this would work for at least one or two amino acids, but it did not seem to. I did not do this comprehensively, but perhaps there are no truly redundant parts of this small binder?
Conclusion
All the top spots on the leaderboard went to Alex Naka, who helpfully detailed his methods in this thread. (A lot of this is similar to what I did above, including using modal!) Anthony Gitter also published an interesting thread on his attempts. I find these kinds of threads are very useful since they give a sense of the tools people are using in practice, including some I had never heard of, like pepmlm and Protrek.
Finally, I made a tree of the 200 designs that Adaptyv is screening (with iPAE <10 in green, <20 in orange, and >20 in red). All the top scoring sequences are EGF-like and cluster together. (Thanks to Andrew White for pointing me at the sequence data). We can look forward to seeing the wet lab results published in a couple of weeks.

Tree of Adaptyv binder designs