Alphafold 3 (AF3) came out in May 2024, and included several major advances over Alphafold 2 (AF2). In this post I will give a brief review of Alphafold 3, and compare the various open and less-open AF3-inspired models that have come out over the past six months. Finally, I will show some results from folding antibody complexes.
Alphafold 3
AF3 has many new capabilities compared to AF2: it can work with small molecules, nucleic acids, ions, and modified residues. It also has arguably a streamlined architecture compared to AF2 (pairformer instead of evoformer, no rotation invariance).
310.ai did a nice review and small benchmark of AlphaFold3 that is worth reading.

The AF3 paper hardly shows any data comparing AF3 to AF2, and is mainly focused on its new capabilities working with non-amino acids. In all cases tested, it performed as well as or exceeded state-of-the-art. For most regular protein folding problems, AF3 and AF2 work comparably well (more specifically, Alphafold-Multimer (AF2-M), the AF2 revision that allowed for multiple protein chains) though for antibodies there is a jump in performance.
Still, despite being an excellent model, AF3 gets relatively little discussion. This is because the parameters are not available so nobody outside DeepMind/Isomorphic Labs really uses it. The open source AF2-M still dominates, especially when used via the amazing colabfold project.
Alphafold-alikes
As soon as AF3 was published, the race was on to reimplement the core ideas. The chronology so far:
| Date | Software | Code available? | Parameters available? | Lines of Python code |
|---|---|---|---|---|
| 2024-05 | Alphafold 3 | ❌ (CC-BY-NC-SA 4.0) | ❌ (you must request access) | 32k |
| 2024-08 | HelixFold3 | ❌ (CC-BY-NC-SA 4.0) | ❌ (CC-BY-NC-SA 4.0) | 17k |
| 2024-10 | Chai-1 | ❌ (Apache 2.0, inference only) | ✅ (Apache 2.0) | 10k |
| 2024-11 | Protenix | ❌ (CC-BY-NC-SA 4.0) | ❌ (CC-BY-NC-SA 4.0) | 36k |
| 2024-11 | Boltz | ✅ (MIT) | ✅ (MIT) | 17k |
There are a few other models that are not yet of interest: Ligo's AF3 implementation is not finished and perhaps not under active development, LucidRains' AF3 implementation is not finished but is still under active development.
It's been pretty incredible to see so many reimplementation attempts within the span of a few months, even if most are not usable due to license issues.
Code and parameter availability
As a scientist who works in industry, it's always annoying to try to figure out which tools are ok to use or not. It causes a lot of friction and wastes a lot of time. For example, I started using ChimeraX a while back, only to find out after sinking many hours into it that this was not allowed.
There are many definitions of "open" software. When I say open I really mean you can use it without checking with a lawyer. For example, even if you are in academia, if the license says the code is not free for commercial use, then what happens if you start a collaboration with someone in industry? What if you later want to commercialize? These are common occurrences.
In some cases (AF3, HelixFold3, Protenix, and Chai-1), they make a server available, which is nice for very perfunctory testing, but precludes testing anything proprietary or folding more than a few structures. If you have the code and the training set, it would cost around $100k to train one of these models (specifically, the Chai-1 and Protenix papers give numbers in this range, though that is just the final run). So in theory there is no huge blocker to retraining. In practice it does not seem to happen, perhaps for license issues.
The specific license matters. Before today, I thought MIT was just a more open Apache 2.0, but apparently there is an advantage to Apache 2.0 around patents! My non-expert conclusion is that unlicense, MIT and Apache are usable, GPL and CC-BY-NC-SA are not.

Which model to choose?
There are a few key considerations: availability; extensibility / support; performance.
1. Availability
In terms of availability, I think only Chai-1 and Boltz are in contention. The other models are not viable for any commercial work, and would only be worth considering if their capabilities were truly differentiated. As far as I know, they are not.
2. Extensibility and support
I think this one is maybe under-appreciated. If an open source project is truly open and gains enough mindshare, it can attract high quality bug reports, documentation, and improvements. Over time, this effect can compound. I think currently Boltz is the only model that can make this claim.
A big difference between Bolt and Chai-1 is that Boltz includes the training code and neural network architecture, whereas Chai-1 only includes inference code and uses pre-compiled models. I only realized this when I noticed the Chai-1 codebase is half the size of the Boltz codebase. Most users will not retrain or finetune the model, but the ability for others to improve the code is important.
To be clear, I am grateful to Chai for making their code and weights available for commercial purposes, and I intend to use the code, but from my perspective Boltz should be able to advance much quicker. There is maybe an analogy to Linux or Blender vs proprietary software.
3. Performance
It's quite hard to tell from the literature who has the edge in performance.
You can squint at the graphs in each paper,
but fundamentally all of these models are AF3-derivatives trained on the same data,
so it's not surprising that performance is generally very similar.
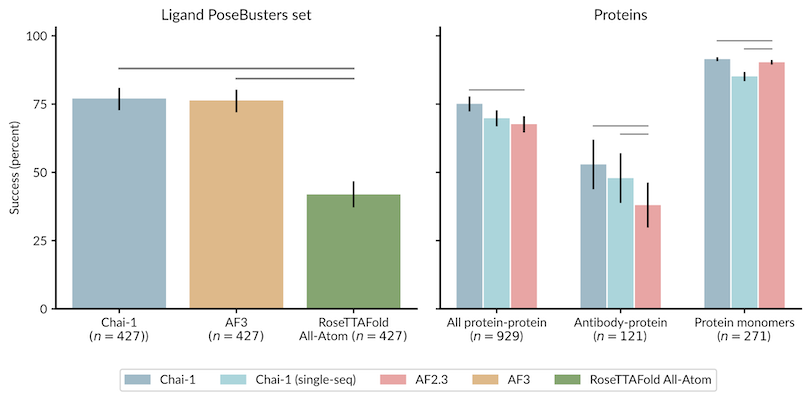
Chai-1 and AF3 perform almost identically

Boltz and Chai-1 perform almost identically

Protenix and AF-3 perform almost identically
Benchmarking performance
I decided to do my own mini-benchmark, by taking 10 recent (i.e., not in any training data) antibody-containing PDB entries and folding them using Boltz and Chai-1.
Both models took around 10 minutes per antibody fold on a single A100 (80GB for Boltz, 40GB for Chai-1). Chai-1 is a little faster, which is expected since it uses ESM embeddings instead of multiple sequence alignments (MSAs). (Note, I did not test Chai-1 in MSA mode, giving it a small disadvantage compared to Boltz.)
Tangentially, I was surprised I could not find a "pdb to fasta" tool that would output protein, nucleic acids, and ligands. Maybe we need a new file format? You can get protein and RNA/DNA from pdb, but it will be the complete sequence of the protein, not the sequence in the PDB file (this may or may not be what you want). Extracting ligands from PDB files is actually very painful since the necessary bond information is absent! The best code I know of to do this is a pretty buried old Pat Walters gist.
Most of the PDBs I tested were protein-only,
one had RNA,
and I skipped one glycoprotein.
I evaluated performance using USalign,
using either the average "local" subunit-by-subunit alignment (USalign -mm 1)
or one "global" all-subunit alignment (USalign -mm 2).
Both models do extremely well when judged on local subunit accuracy,
but much worse for global accuracy — sadly this is quite relevant for an antibody model!
It appears that these models well understand how antibodies fold, but not how they bind.
Conclusions
On my antibody benchmark, Boltz and Chai-1 perform eerily similar, with a couple of cases where Boltz wins out. That, combined with all the data from the literature, makes the conclusion straightforward, at least for me. Boltz performs as well as or better than any of the models, has a clean, complete codebase with relatively little code, is hackable, and is by far the most open model. I am excited to see how Boltz progresses in 2025!
Technical details
I ran Boltz and Chai-1 on modal using my biomodals repo.
modal run modal_boltz.py --input-faa 8zre.fasta --run-name 8zre
modal run modal_chai1.py --input-faa 8zre.fasta --run-name 8zre
Here is a folder with all the pdb files and images shown below.
Addendum
On BlueSky, Diego del Alamo notes that Chai-1 outperformed Boltz in a head-to-head of antibody–antigen modeling.
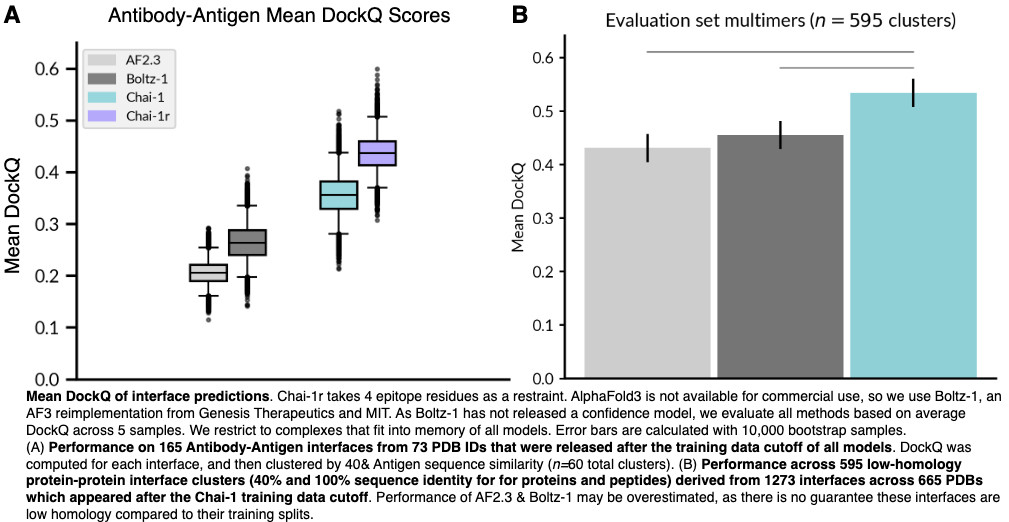
On linkedin, Joshua Meier
(co-founder Chai Discovery) recommended running Chai-1 with msa_server turned on,
to make for a fairer comparison.
I reran the benchmark with Chai-1 using MSAs, and it showed improvements
in 8ZRE (matching Boltz) and 9E6K (exceeding Boltz.)
I think it is still fair to say that the results are very close.
| Complex | Boltz | Chai-1 | ||||
|---|---|---|---|---|---|---|
| 9CIA: T cell receptor complex |
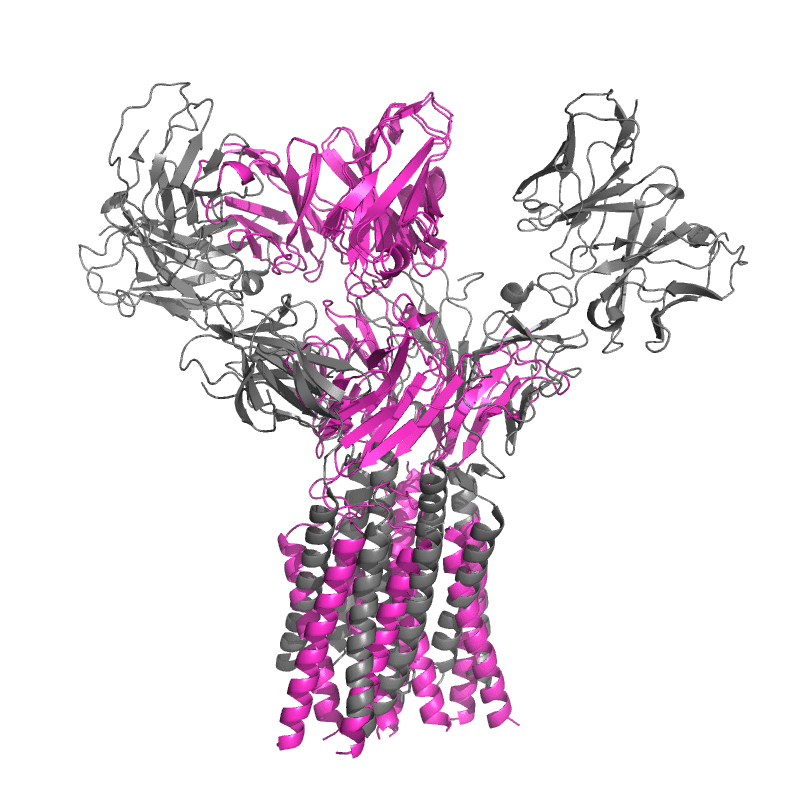
|
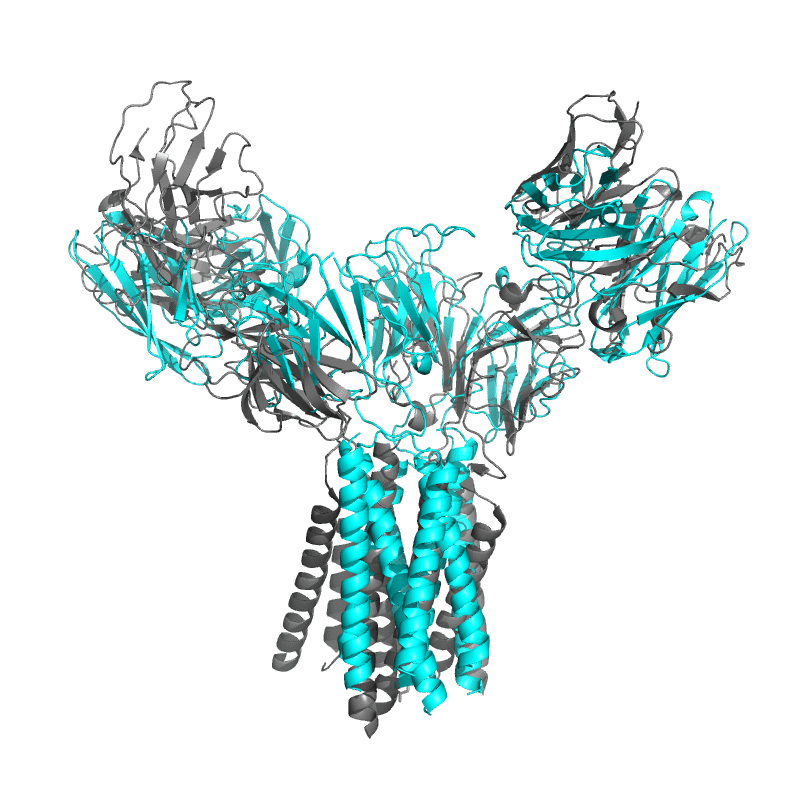
|
||||
| 8ZRE: HBcAg-D4 Fab complex |
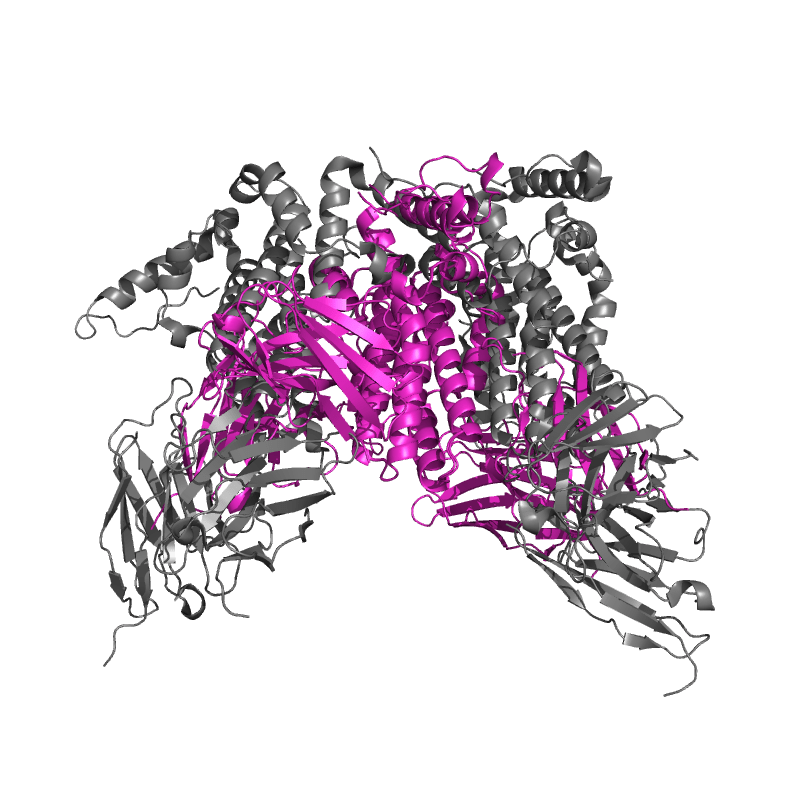
|
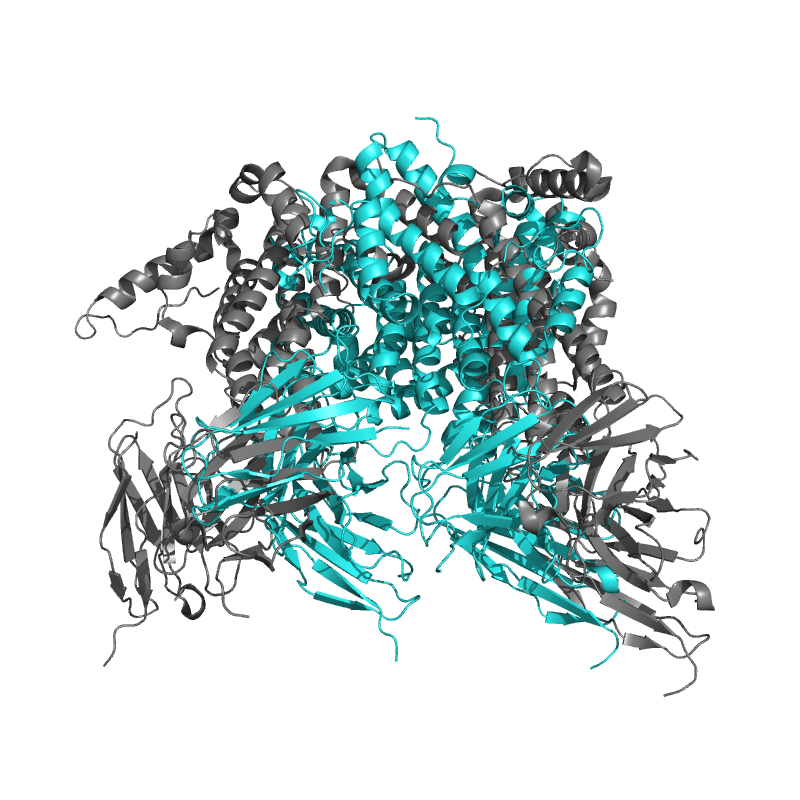
|
||||
| 9DF0: PDCoV S RBD bound to PD41 Fab (local refinement) |
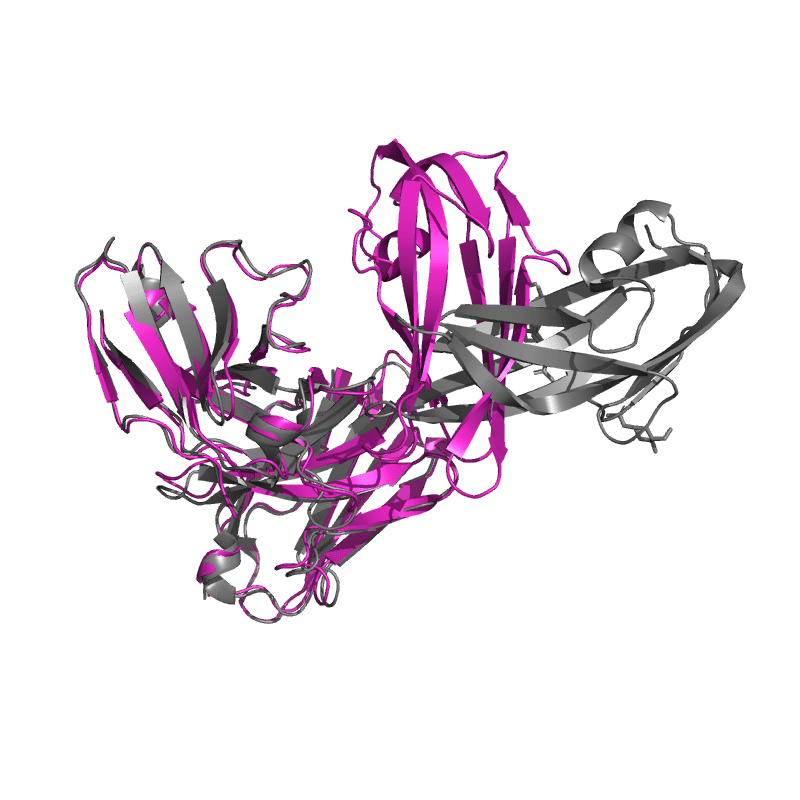
|
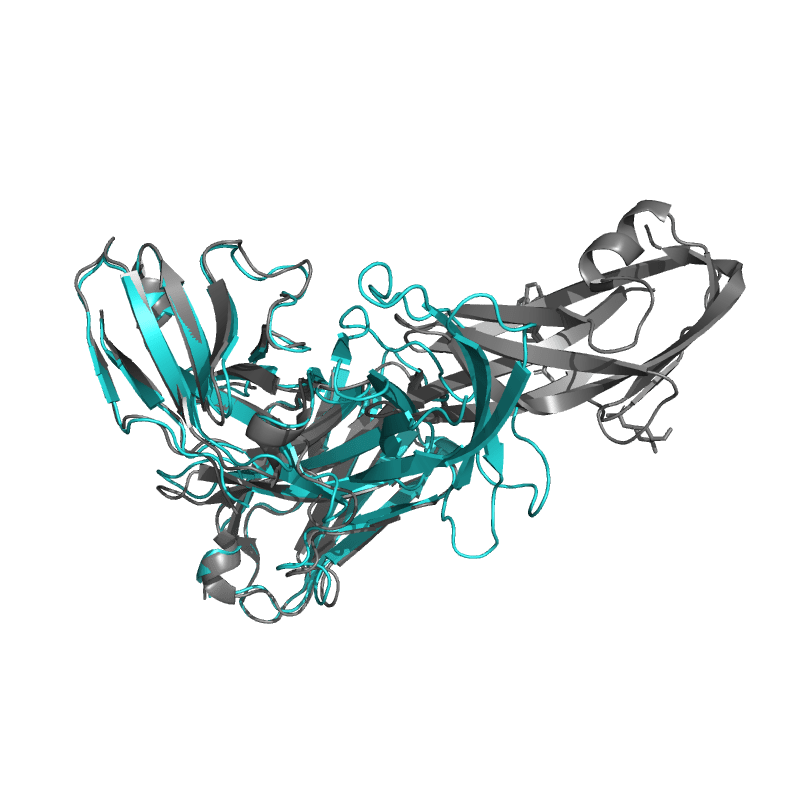
|
||||
| 9CLP: Structure of ecarin from the venom of Kenyan saw-scaled viper in complex with the Fab of neutralizing antibody H11 |
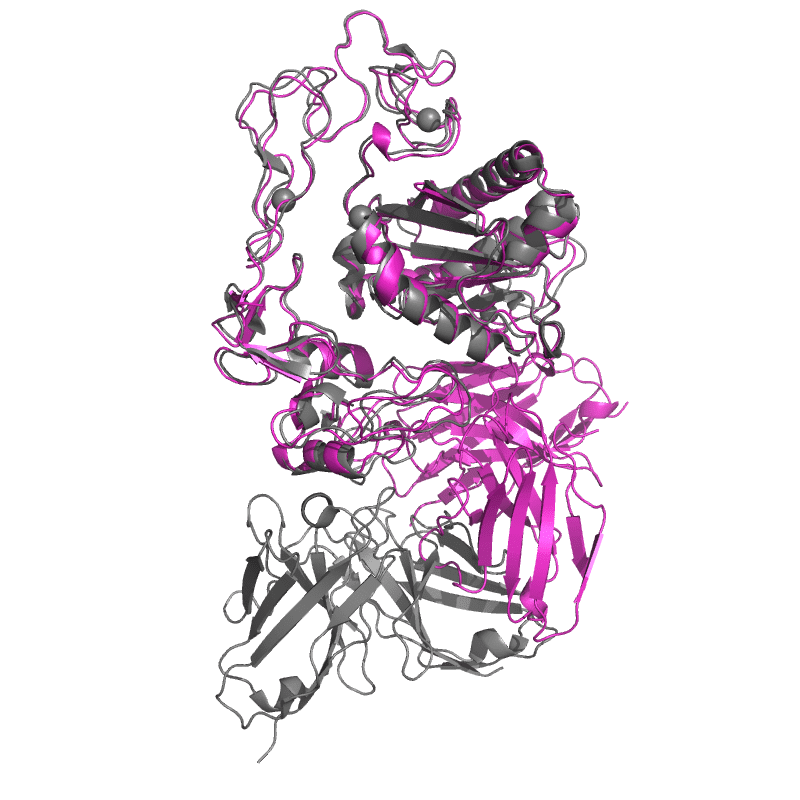
|

|
||||
| 9C45: SARS-CoV-2 S + S2L20 (local refinement of NTD and S2L20 Fab variable region) |
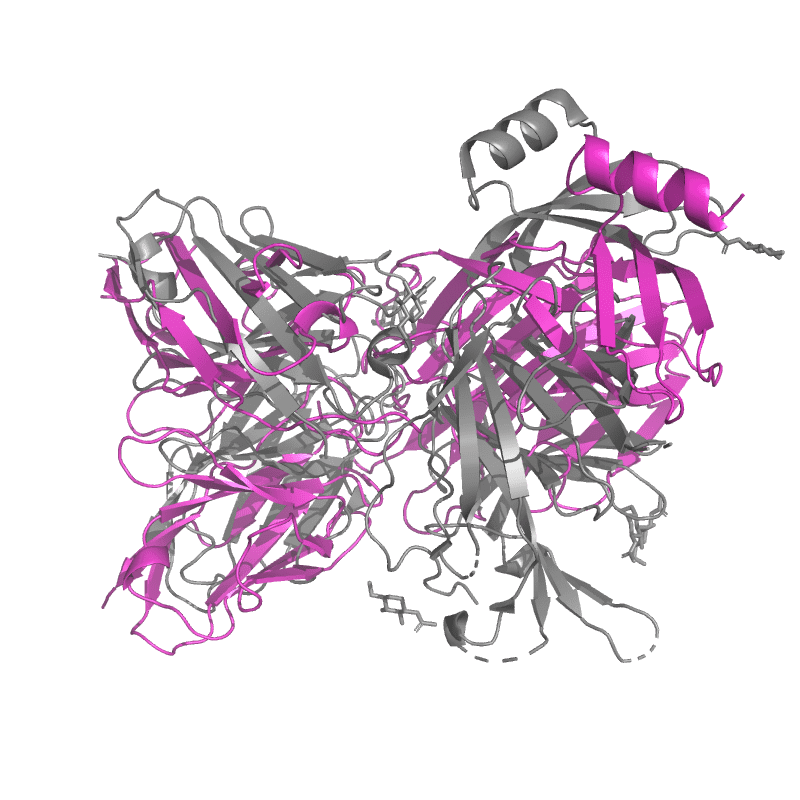
|

|
||||
| 9E6K: Fully human monoclonal antibody targeting the cysteine-rich substrate-interacting region of ADAM17 on cancer cells. |
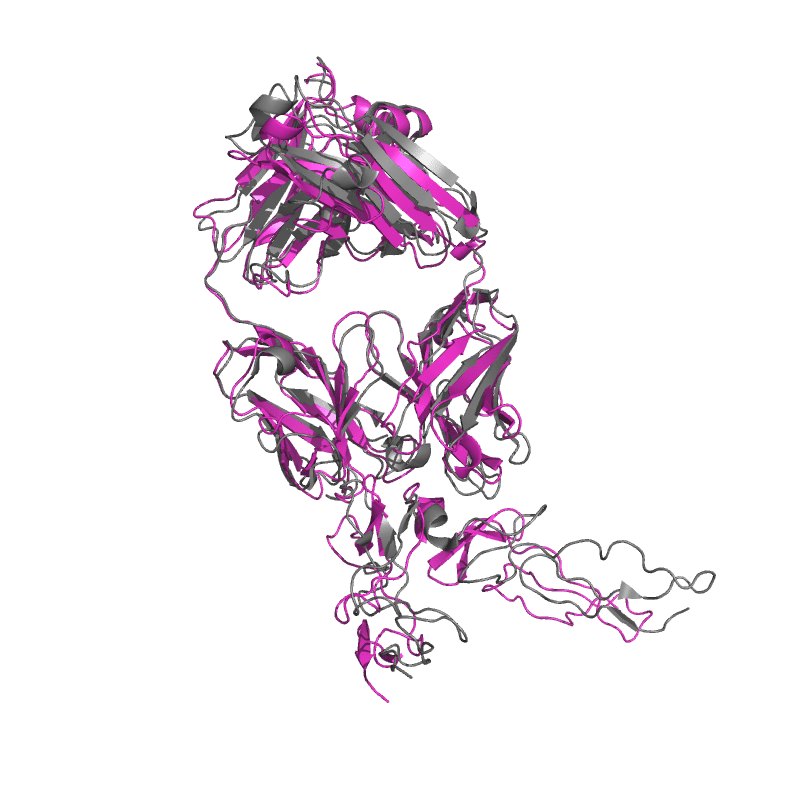
|
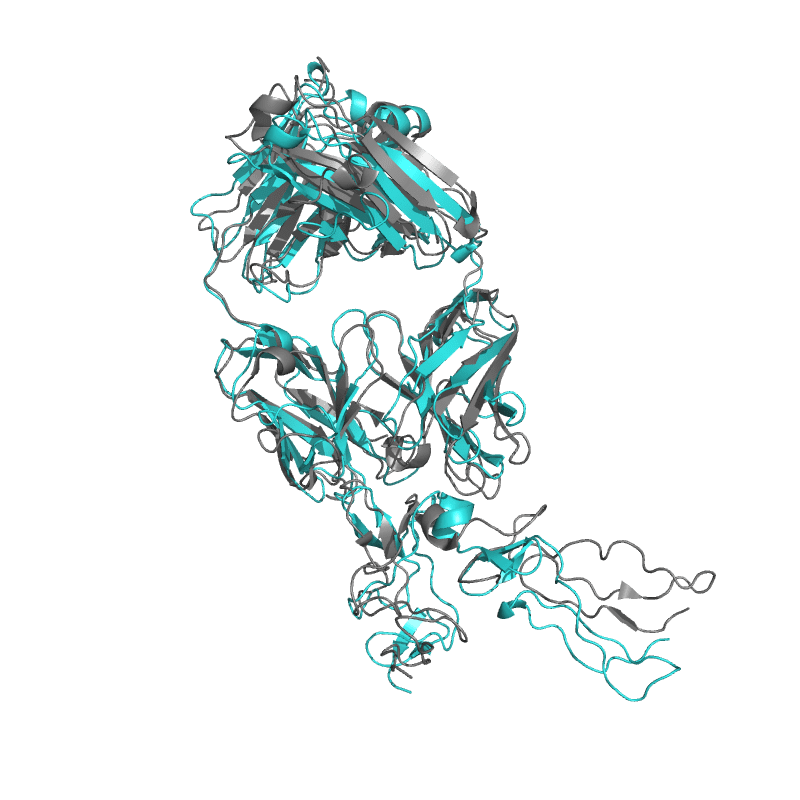
|
||||
| 9CMI: Cryo-EM structure of human claudin-4 complex with Clostridium perfringens enterotoxin, sFab COP-1, and Nanobody |
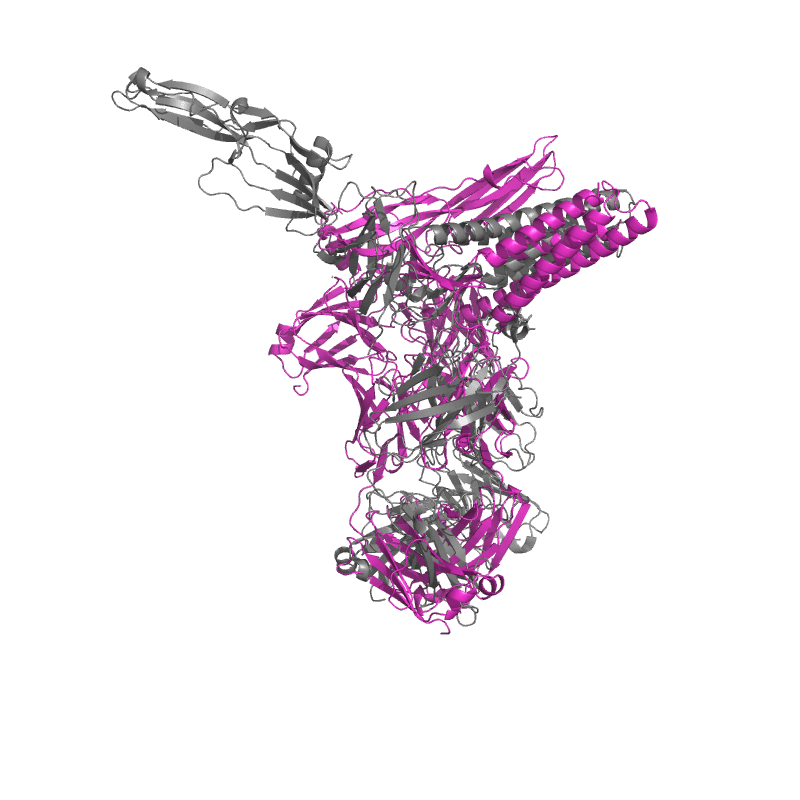
|

|
||||
| 9CX3: Structure of SH3 domain of Src in complex with beta-arrestin 1 |

|
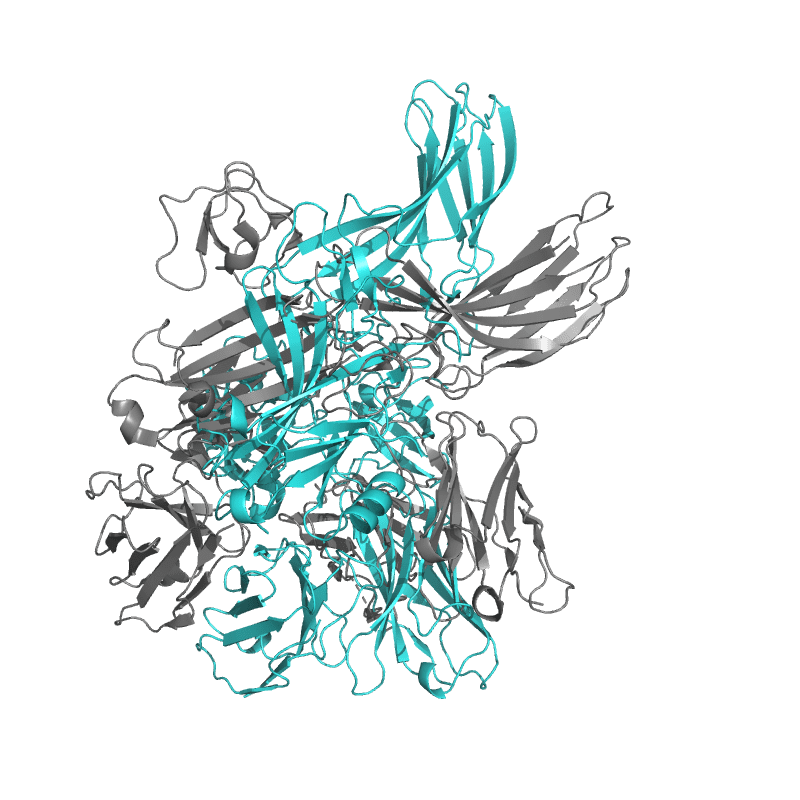
|
||||
| 9DX6: Crystal structure of Plasmodium vivax (Palo Alto) PvAMA1 in complex with human Fab 826827 |
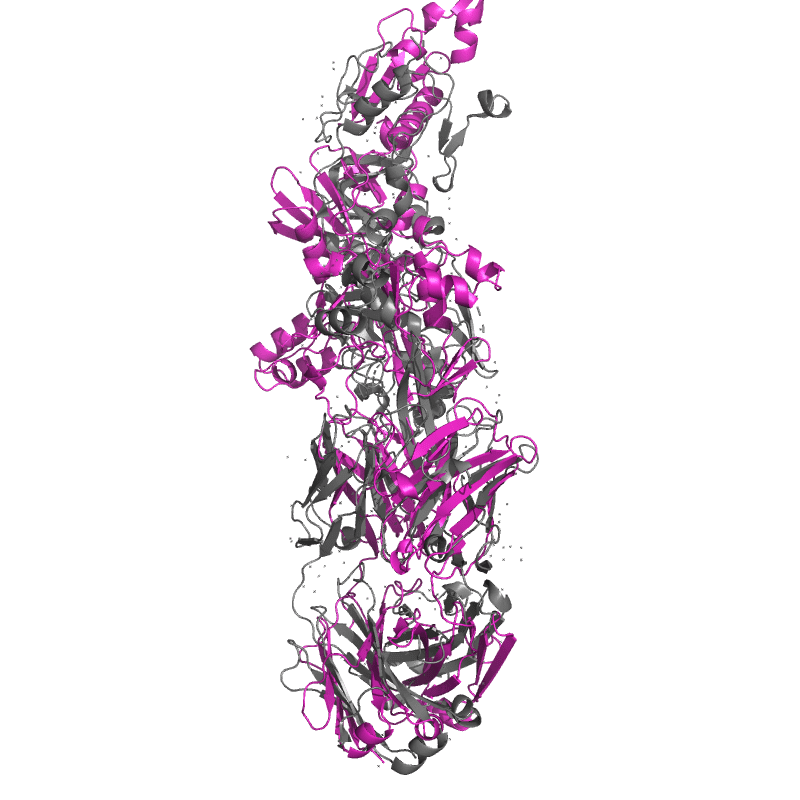
|
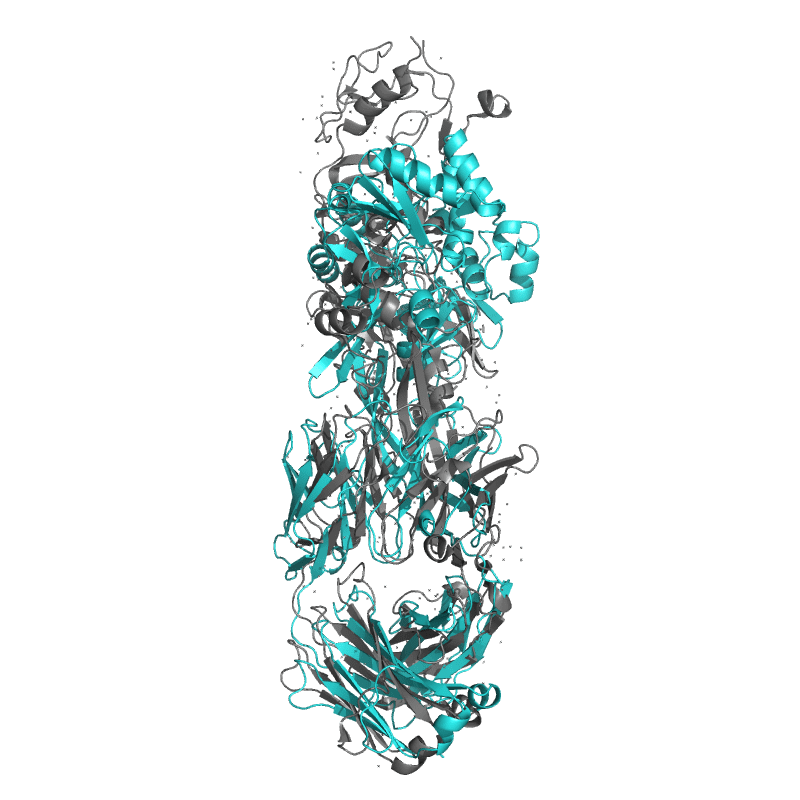
|
||||
| 9DN4: Crystal structure of a SARS-CoV-2 20-mer RNA in complex with FAB BL3-6S97N . |

|
|