In this post I'll describe how to sequence a human genome at home, something that's only recently become possible.
The protocol described here is not necessarily the best way to do this, but it's what has worked best for me. It costs a few thousand dollars in equipment to get started, but the (low-coverage) sequencing itself requires only $150, a couple of hours of work, and almost no lab skills.
What does it mean to sequence a human genome?
First, it's useful to explain some terms: specifically, to differentiate a regular reference-based genome assembly from a de novo genome assembly.
Twenty years or so ago, the Human Genome Project resulted in one complete human genome sequence (actually combining data from several humans). Since all humans are 99.9%+ identical genetically, this reference genome can be used as a template for any human. The easiest way to sequence a human genome is to generate millions of short reads (100-300 base-pairs) and aligning them to this reference.
The alternative to this reference-based assembly is a de novo assembly, where you figure out the genome sequence without using the reference, by stitching together overlapping sequencing reads. This is much more difficult computationally (and actually impossible if your reads are too short), but the advantage is that you can potentially see large differences compared to the reference. For example, it's not uncommon to have some sequence in a genome that is not present in the reference genome, so-called structural variants.
There are also gaps in the reference genome, especially at the ends and middle of chromosomes, due to highly repetitive sequence. In fact, the first full end-to-end human chromosome was only sequenced last year, thanks to ultra-long nanopore reads.
For non-human species, genome assembly is usually de novo, either because the genomes are small and non-repetitive (bacteria), or there is no reference (newly sequenced species).
SNP chip
The cheapest way to get human genome sequence data is with a SNP chip, like the 23andMe chip. These chips work by measuring variation at specific, pre-determined positions in the genome. Since we know the positions that commonly vary in the human genome, we can just examine a few hundred thousand of those positions to see most of the variation. You can also accurately impute tons of additional variants not on the chip. The reason this is "genotyping" and not "sequencing" is that you don't get a contiguous sequence of As, Cs, Gs, and Ts. The major disadvantage of SNP chips is that you cannot directly measure variants not on the chip, so you miss things, especially rare and novel variants. On the other hand, the accuracy for a specific variant of interest (e.g., a recessive disease variant like cystic fibrosis ΔF508) is probably higher than from a sequenced genome.
Short-read sequencing
Short-read sequencing is almost always done with Illumina sequencers, though other short-read technologies are emerging. These machines output millions or billions of 100-300 base-pair reads that you can align to the reference human genome. Generally, people like to have on average 30X coverage of the human genome (~100 gigabases) to ensure high accuracy across the genome.
Although you can read variants not present on a SNP chip, this is still not a complete genome: coverage is not equal across the genome, so some regions will likely have too low coverage to call variants; the reference genome is incomplete; some structural variants (insertions, inversions, repetitive regions) cannot be detected with short reads.
Long-read sequencing
The past few years have seen single-molecule long-read sequencing develop into an essential complement and sometimes credible alternative to Illumina. The two players, Pacific Biosciences and Oxford Nanopore (ONT) are now mature technologies.
The big advantage of these technologies is that you get reads much longer than 300bp — from low thousands up to megabases on ONT in extreme examples — so assembly is much easier. This enables de novo assembly, and is especially helpful with repetitive sequence. For this reason, long-read sequencing is almost essential for sequencing new species, especially highly repetitive plant genomes.
Sounds great! Why do people still use Illumina then? The per-base accuracy and per-base cost of Illumina is still considerably better than these competitors (though ONT's PromethION is getting close on price).
One huge advantage that ONT has over competitors is that the instrument is a fairly simple solid-state device that reads electrical signals from the nanopores. Since most of the technology is in the consumable "flow-cell" of pores, the instruments can be tiny and almost free to buy.
Instead of spending $50k-1M on a complex machine that requires a service contract, etc., you can get a stapler-sized MinION sequencer for almost nothing, and you can use it almost anywhere. ONT have also done a great job driving the cost per experiment down, especially by releasing a lower-output flow-cell adaptor called the flongle. Flongle flow-cells only cost $90 per flow-cell, and produce 100 megabases to >1 gigabase of sequence.
There is a great primer on how nanopore sequencing works at nanoporetech.com.
Nanopore Sequencing Equipment
(Note, to make this article stand alone, I copied text from my previous home lab blogpost.)
Surprisingly, you don't actually need much expensive equipment to do nanopore sequencing.
In my home lab, I have the following:
- Three calibrated pipettes for $45 each and boxes of three sizes of pipette tips for $15 each from the-odin.com.
- An Eppendorf 5415C centrifuge from ebay for around $300. DNA extraction requires a centrifuge that can spin at >=12k rpm, which means a large-ish lab-grade centrifuge.
- A mini-centrifuge for $45 from the-odin.com. They also sell a nice 10k rpm version for $125.
- Two sous vides for water baths, e.g. this Anova sous vide for $99 from amazon.
- A vortex mixer for $80 from amazon.
- A MinION nanopore sequencer. Unfortunately, you do not get a MinION with a flongle starter pack ($1,460). The only way I know of to get a new one is in a MinION starter pack for $1,000, which includes one MinION flow-cell, worth approximately $1000.
- A refrigerator that does not have a defrost cycle. I bought a small mini-fridge from amazon for $150 so my lab stuff would not be in the kitchen fridge.
- I already had a chest freezer that does not have a defrost cycle.
Optional equipment:
- A wireless fridge thermometer. This was only $25 and it works great! It's useful to be able to keep track of temperatures in your fridge or freezer. Some fridges can get cold enough to freeze, which is deadly for flow-cells.
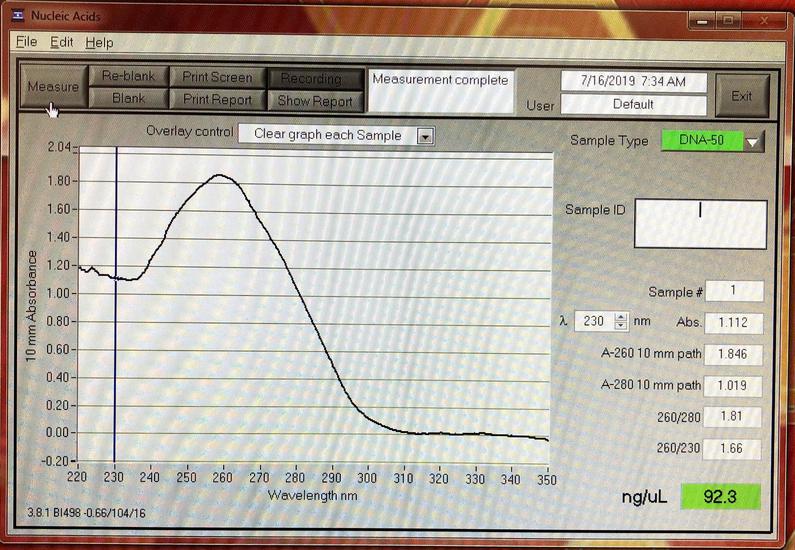
- A GeneQuant to check the quality of DNA extractions. A 20 year old machine cost me about $150 on ebay. It's a useful tool, but does require quite a lot of sample (I use 200 µl). I wrote a bit more about it here.



Protocol Part One: extracting DNA
The first step in sequencing is DNA extraction (i.e., isolating DNA from biological material). I use a Zymo Quick-DNA Microprep Plus Kit, costing $132. It's 50 preps, so a little under $3 per prep. There are other kits out there, like NEB's Monarch, but these are harder to buy (requiring a P.O., or business address).
The Zymo kit takes "20 minutes" (it takes me about 40 minutes including setting up). It is very versatile: it can work with "cell culture, solid tissue, saliva, and any biological fluid sample". This prep is pretty easy to do, and all the reagents except Proteinase k are just stored at room temperature. They claim it can recover >50kb fragments, and anecdotally, this is the maximum length I have seen. That is far from the megabase-long "whale" reads some labs can achieve, but those preps are much more complex and time-consuming. Generally speaking, 10kb fragments are more than long enough for most use-cases.
Protocol Part Two: library prep
Library prep is the process of preparing the DNA for sequencing, for example by attaching the "motor protein" that ratchets the DNA through the pore one base at a time. The rapid library prep (RAD-004) is the simplest and quickest library prep method available, at $600 for 12 preps ($50 per prep).
Library prep is about as difficult as DNA extraction, and takes around 30 minutes. There are some very low volumes involved (down to 0.5µl, which is as low as my pipettes go), and you need two to use two water bath temperatures, but overall it's pretty straightforward.
The total time from acquiring a sample to beginning sequencing could be as little as 60-90 minutes. You do pay for this convenience in lower read lengths and lower throughput though.
The Data
The amount of data you can get from ONT/nanopore varies quite a lot. There is a fundamental difference between Illumina and nanopore in that nanopore is single-molecule sequencing. With nanopore, each read represents a single DNA molecule traversing the pore. With Illumina, a read is an aggegated signal from many DNA molecules (which contributes to the accuracy).
So, nanopore is really working with the raw material you put in. If there are contaminants, then they can jam up the pores. If there are mostly short DNA fragments in the sample, you will get mostly short reads. Over time, the pores degrade, so you won't get as much data from a months-old flow-cell as a new one.
Using the protocol above, I have been able to get around 100-200 megabases of data from one flongle ($1 per megabase!). There are probably a few factors contributing to this relatively low throughput: the rapid kit does not work as well as the more complex ligation kit; I don't do a lot of sequencing, so the protocol is certainly executed imperfectly; my flow-cells are not always fresh.
For a human sample, 100 megabases is less than a 0.1X genome, which raises the fair question of why you would want to do that? Today, the answer is mainly just because you can. You could definitely do some interesting ancestry analyses, but it would be difficult to validate without a reference database. gencove also has several good population-level use-cases for low-pass sequencing.
The next step up from a flongle is a full-size MinION flow-cell, which runs on the same equipment and uses the same protocol, but costs around $900, and in theory can produce up to 42 gigabases of sequence. This would be a "thousand dollar genome", though the accuracy is probably below what you would want for diagnosic purposes. In a year or two, I may be able to generate a diagnostic-quality human genome at home for around $1000, perhaps even a decent de novo assembly.
Comment