The amyloid hypothesis
There have been a lot of results coming out from Alzheimer's trials recently, and a lot of discussion about the "amyloid hypothesis" and its role in the disease. In this post I'll review some of the evidence, and see how it relates to data from recent AD trial results from Merck and Biogen/Eisai. I mainly reference three good reviews that cover most of the basic facts and arguments around the amyloid hypothesis. Much of my additional data is from AlzForum, a fantastic resource for Alzheimer's news.
- The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics (2011)
- The amyloid hypothesis of Alzheimer's disease at 25 years (2016)
- The case for rejecting the amyloid cascade hypothesis (2015)
The basics
A simplified model of the amyloid hypothesis is that the cell-surface protein APP (Amyloid Precursor Protein) gets cleaved by BACE1 and γ-secretase and released as a 42 amino acid peptide, Aβ42; Aβ42 forms oligomers, then extracellular plaques in the brain; these oligomers and/or plaques somehow lead to intracellular Tau tangles which cause neuronal death.
One big question here is whether it's the plaques or oligomers that are the main trigger:
Several similar studies suggest that Aβ — particularly soluble oligomers of Aβ42 (Shankar et al, 2008) — can trigger AD‐type tau alterations
This model is nicely summarized by a diagram from NRD:
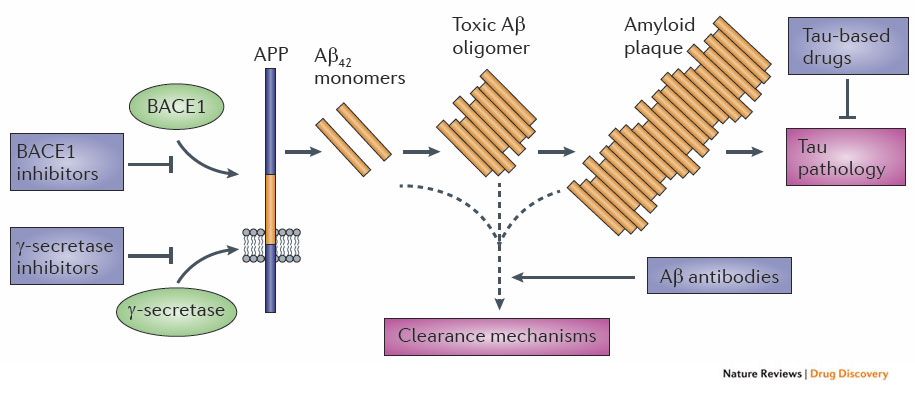
As the diagram shows, the obvious drug targets are γ-secretase and BACE1 (to stop Aβ42 production), Aβ42 monomers/oligomers/plaques (to reduce plaque formation), Tau (to prevent Tau tangles).
There have been drugs targeting all of these processes. None have been successful:
- gamma-secretase: semagacestat (Lilly), a gamma-secretase inhibitor, failed Phase III in 2010
- BACE1: lanabecestat (Lilly) failed Phase III in 2018; verubecestat (Merck) failed Phase III in 2017 and 2018
- Tau: LMTM (TauRX) failed Phase III in 2016; AbbVie's anti-Tau antibody is still in Phase II.
- Aβ42: solanezumab (Lilly) failed Phase III in 2016 and 2018; bapineuzumab (Pfizer) failed Phase III in 2012; crezenumab (Roche) is still in Phase III; aducanumab (Biogen) is still in Phase III.
The only approved drugs for Alzheimer's are fairly ineffectual cholinesterase inhibitors (and an accompanying NMDA receptor inhibitor). These drugs are usually thought of more as symptom relief than treatment.
Aβ42 Antibodies
Why do drug companies keep making Aβ42 antibodies after so many failures? In fact, there is quite a bit of variability in what these antibodies actually do. Ryan Watts, now CEO of Denali Therapeutics, gave an interview with AlzForum back in 2012 where he explained the difference between Genentech's crezenumab and other Aβ42 antibodies.
Q: How is crenezumab different from the other Aβ antibodies that are currently in Phase 2 and 3 trials?
A: We have a manuscript under review that describes its properties. Basically, crenezumab binds to oligomeric and fibrillar forms of Aβ with high affinity, and to monomeric Aβ with lower affinity. By comparison, solanezumab binds monomeric Aβ, and gantenerumab binds aggregated Aβ, as does bapineuzumab. Crenezumab binds all forms of the peptide. Crenezumab is engineered on an IgG4 backbone, which allows it to activate microglia just enough to promote engulfment of Aβ, but not so strongly as to induce inflammatory signaling through the p38 pathway and release of cytokines such as tumor necrosis factor α. Crenezumab is the only IgG4 anti-Aβ antibody in clinical development that I am aware of. We have not seen vasogenic edema in our Phase 1 trials, which was the first main hurdle for us to overcome.
Biogen describes the MOA of their aducanumab antibody like this:
Aducanumab is thought to target aggregated forms of beta amyloid including soluble oligomers and insoluble fibrils which can form into amyloid plaque in the brain of Alzheimer’s disease patients.
Denali Therapeutics
As an aside, Denali is not working on an Aβ42 inhibitor (perhaps for IP reasons since Ryan Watts was heavily involved in the development of crezenumab). Apart from their novel RIPK1 program, they are still pursuing BACE1 and Tau.
Our lead RIPK1 product candidate, DNL747, is a potent, selective and brain penetrant small molecule inhibitor of RIPK1 for Alzheimer’s disease and ALS. Microglia are the resident immune cells of the brain and play a significant role in neurodegeneration. RIPK1 activation in microglia results in production of a number of pro-inflammatory cytokines that can cause tissue damage.
Our three antibody programs are against known targets including aSyn, TREM2 and a bi-specific therapeutic agent against both BACE1 and Tau. Our BACE1 and Tau program is an example of combination therapy, which we believe holds significant promise in developing effective therapies in neurodegenerative diseases.
How does amyloid cause disease?
By one definition, the amyloid hypothesis "posits that the deposition of the amyloid-β peptide in the brain is a central event in Alzheimer's disease pathology". There are several ways that amyloid could cause AD. This diagram from a 2011 NRD review shows three options:

- Aβ trigger: Aβ triggers the disease once it reaches a threshold, and once it starts, reducing Aβ levels does not help
- Aβ threshold: Aβ triggers the disease once it reaches a threshold, but reducing Aβ levels back below the threshold does help
- Aβ driver: Aβ causes Alzheimer's, and reducing Aβ levels at any time should ameliorate disease
Simplifying, if the Aβ trigger model is correct, then we don't expect anti-Aβ42 antibodies to work, except perhaps preventatively. If the Aβ driver model is correct, then these antibodies should work, at least partially.
From the same review:
A strong case can be made that the deposition of amyloid-β in the brain parenchyma is crucial for initiating the disease process, but there are no compelling data to support the view that, once initiated, the disease process is continuously driven by or requires amyloid-β deposition.
For this reason, after Aβ42 antibody trials fail, the stock answer from pharma is that they need to begin treatment earlier. Of course, the earlier you treat, the longer the trial takes, and the more you need new amyloid detection technologies like Florbetavir/PET to see what's going on. So it's probably natural that there is a gradual transition to ever earlier interventions and longer trials, even though this can also seem like excuse-making.
Evidence for the amyloid hypothesis
Despite all the failed drugs and holes in our understanding, the amyloid hypothesis remains durable due to the weight of evidence in its corner.
APP
Mutations in APP both cause and prevent Alzheimer's. Half of people with trisomy 21 (or any APP duplication, it seems) develop AD by the time they reach their fifties.
A protective variant found in APP also points to a causal relationship, and therapeutic potential (see Robert Plenge on allelic series).
We found a coding mutation (A673T) in the APP gene that protects against Alzheimer's disease and cognitive decline in the elderly without Alzheimer's disease. This substitution is adjacent to the aspartyl protease β-site in APP, and results in an approximately 40% reduction in the formation of amyloidogenic peptides in vitro. Carriers are about 7.5 times more likely than non-carriers to reach the age of 85 without suffering major cognitive decline
A cryoEM structure of Aβ42 fibril from 2017 gives us structural evidence for why APP mutations should be protective or damaging, suggesting that APP's effect on AD is via amyloid/Aβ42.

APOE
The APOE e4 allele strongly predisposes people to Alzheimer's. It's one of the strongest genetic associations known, besides Mendelian diseases. In 2018, Yadong Huang's team at the Gladstone Institiute used iPSCs to investigate the mechanism. Confusingly, they found that APOE is independently associated with both Aβ42 and Tau.
"ApoE4 in human neurons boosted production of Aβ40 and Aβ42"
"It does not do that in mouse neurons. Independent of its effect on Aβ, ApoE4 triggered phosphorylation and mislocalization of tau."
"Based on these data, we should lower ApoE4 to treat AD"
This research may also help explain why mouse models of Alzheimer's have often been misleading.
Other evidence
- Mutations in PSEN1 and PSEN2 (components of gamma-secretase) cause Alzheimer's.
- Other diseases are caused by mutations in amyloid-forming proteins. For example, there is a mutation that enables IAPP to form amyloid, which causes Familial British Dementia.
In ALS, the aggregated form of SOD1 may be protective and the soluble form disease-causing.
The formation of large aggregates is in competition with trimer formation, suggesting that aggregation may be a protective mechanism against formation of toxic oligomeric intermediates.
Criticism of the amyloid hypothesis
The main criticism of the antibody hypothesis is that we have been testing anti-amyloid drugs — especially antibodies against Aβ42 — for a long time now, and none of them have had any effect on disease progression.
Derek Lowe (and many of his commenters) has written especially skeptically on his blog:
Eli Lilly remains committed to plunging through this concrete wall headfirst. [...] our gamma-secretase inhibitor completely failed in 2010. Then we took our antibody, solanezumab into Phase III trials that failed in 2012. And found out in 2013 that our beta-secretase inhibitor failed.
Morgan Sheng, VP of Neuroscience at Genentech, is much more positive. In a recent interview in NRD he said:
Let me start by saying that I fully believe in the amyloid hypothesis, and I think it’s going to be vindicated completely within years. [...] phase III results from drugs like Eli Lilly’s solanezumab suggest these agents sort of work; they just don’t work very well
It seems like targeting Tau is an acceptable strategy to amyloid hypothesis skeptics because it's not targeting Aβ42, even though it's still part of the standard amyloid hypothesis model. Drugs that are based on the "amyloid hypothesis" and drugs that work by trying to reduce amyloid tend to get conflated in a confusing way.
Evidence against the amyloid hyothesis
Here I am mainly summarizing from a 2015 review. In this review, the author mainly disputes the "linear story" of the amyloid hypothesis and not the fact that Aβ plays some kind of role in AD.
-
Many people have plaque but no disease.
The existence of this group of individuals (healthy, but amyloid positive) is a substantial challenge to the amyloid cascade hypothesis. It is clearly possible to have amyloid deposits without dementia; therefore amyloid is not sufficient to cause disease.
Such individuals are not rare; rather, they account for a quarter to a third of all older individuals with normal or near-normal cognitive function.
-
Anti-Aβ42 antibodies can reduce plaque without alleviating the disease.
The second test of the amyloid cascade hypothesis has also been done: amyloid has been removed from the brains of individuals with AD and from mice with engineered familial forms of the disease. Here the tests have been less definitive and the evidence is mixed.
-
Other drugs that should work (beta-secretase inhibitors, gamma-secretase inhibitors, BACE1 inhibitors, Tau inhibitors) don't appear to work.
-
Mutations in the Tau gene can cause dementia without plaques forming, so amyloid is not a necessary step in the process.
-
We do not understand AD pathology well. For example, what are the toxic species of Aβ and Tau? What is the connection between Aβ and tangle pathology? Do Tau tangles spread between neurons like prions?
-
There are other possible causes of AD. For example, certain infections could be causative.
Infection
Recent work showing an association between herpes virus and Alzheimer's could be thought of as supporting or disputing the amyloid hypothesis. In this model, the virus "seeds" amyloid plaque formation, which then sequesters the virus. The idea that amyloid plaques are protective is not entirely new, beginning with the "bioflocculant hypothesis" for Aβ, published in 2002.
[Robinson and Bishop] posited that Aβ’s aggregative 332 properties could make it ideal for surrounding and sequestering pathogenic agents in the brain
If herpes causes AD, then we'd expect to see evidence in epidemiological datasets. Both herpes infection and periodontitis appear to be associated with AD risk. Further, antiherpetic medications appear to reduce the risk of AD. A lot more could be done here with a large database of phenotypic information, like UK biobank...
Relatedly, a Bay Area company, Cortexyme, recently raised $76M to pursue an AD drug against a bacterial protease found in plaques.
Recent news
So what about the recent trial results? There were two major trials with new results this year: Merck's BACE1 inhibitor, verubecestat, and Biogen/Eisai's anti-Aβ42 antibody, BAN2401. Meanwhile the trial design for Biogen's aducanumab is being tweaked — not a good sign generally — and there should be new data on that later this year.
Verubecestat
After failing a Phase III in 2017 verubecestart had more bad news last month:
Treatment with verubecestat reduced the concentration of Aβ-40 and Aβ-42 in cerebrospinal fluid by 63 to 81%, which confirms that the drug had the intended action of reducing Aβ production. In the PET amyloid substudy, treatment with verubecestat reduced total brain amyloid load by a modest amount; the mean standardized uptake value ratio was reduced from 0.87 at baseline to 0.83 at week 78 in the 40-mg group. These results suggest that lowering Aβ in the cerebrospinal fluid is associated with some reduction in brain amyloid.
Notably, despite the drug working as intended, the reduction in brain amyloid was minimal.
Hence, some people claim that amyloid removal has not been tested:

Merck's Aβ42 antibody, BAN2401
New Phase II results
for Merck's soluble protofibril antibody,
BAN2401, were just released in July 2018.
The results were hotly disputed because while the Bayesian analysis failed to show an effect,
an alternative p-value based analysis (ANCOVA) showed positive results.
I don't know exactly what the differences between the analyses were,
but generally you would hope for agreement between the two,
unless the effect was pretty marginal or just not real.
The data pulled out in the tweet below shows how strange this situation is.

Given the ambivalent nature of the result, naturally some saw it as positive news, since there was at least something, while skeptics saw the opposite.
Aducanumab
Aducanumab often seems like the most promising anti-Aβ antibody, and maybe the last chance for anti-Aβ antibodies to prove themselves. Back in 2015, Aducanumab showed some promising Phase Ib results. (I even wrote about it).
“They’re the most striking data we have seen with anything, period,” says [an AD trialist]
However, since then the many related trial failures, plus Biogen changing the trial design due to "variability", have left many people pessimistic. Perhaps BAN2401's recent results, however unsatisfying, show that an Aβ inhibitor is not just doomed to show no effect.
Conclusions
There doesn't actually seem to be much controversy about whether amyloid has a role in Alzheimer's; the genetic evidence is especially hard to dispute. I think the disagreement is more whether reducing Aβ plaques (or oligomers) can treat or prevent Alzheimer's. If the plaque is protective, then it's possible that reducing plaque may even worsen the disease.
There are also still plenty of unanswered disease mechanism questions, like whether it's oligomers or plaques that are causative, how Tau tangles cause neuronal death, and how tangles spread from neuron to neuron. Also, a 2018 paper suggests that Tau's function is the opposite of what we thought: instead of stabilizing microtubules, it keeps them dynamic.
One obvious question is why are there not more Tau-based drugs? Tau pathology is not a new idea and Tau's causal relationship with dementia is one of the least controversial parts of the AD story. In fact, there are now at least five Phase I trials underway, so these drugs might just be lagging behind Aβ42 antibodies by a few years. Certainly, Tau tangles being intracellular and in the brain makes drug development more complicated.
"Anti-tau antibodies don’t enter neurons and they don’t bind intracellular tau. We’ve invested a lot of careful rigorous work to try and understand this and I hope that the field will agree that we can put to rest that question"
(Crossing the blood-brain barrier is a problem for almost all AD drugs and especially antibodies — an interesting rule of thumb is that about 0.1% of antibody gets into the brain.)
Despite all the failures, I think the story is coming together and I'm pretty optimistic. We haven't actually tried that many ways of attacking the disease. I think that reducing plaque and/or oligomers very early could still work — mainly because we have seen the "drug" APP A673T working — meanwhile, reducing Tau tangles is arguably the most promising avenue of intervention, and it is yet to be properly tested.