There is an interesting project currently running at Ora Biomedical, a longevity-focused biotech, called the Million Molecule Challenge. The idea is to test as many molecules as possible in a C. elegans longevity assay to look for something better than rapamycin. Ora claims to have already found a better MTOR inhibitor in their initial screens.
Individuals can sponsor testing one of 2720 FDA-approved compounds for $100.

The Million Molecule Challenge
This seemed like a good application for the LLM code from my previous blogpost. The idea is as follows: for each of the 2720 compounds, search PubMed for abstracts around longevity and aging; assess the evidence with an LLM; give each a score from 0-10.
LLM
I decided to try Claude this time instead of GPT-4, having heard good things about Claude Haiku. Haiku is extremely inexpensive at 25c per million tokens input. By comparison GPT-4 is $10 per million tokens. Anthropic also has the more advanced Sonnet ($3/million) and Opus ($15/million) models.
I wanted to try using Haiku, but also run enough compounds through Sonnet and Opus to validate they all gave approximately the same quality answers. In practice, I ran out of Haiku tokens too quickly (my limit was 1M tokens per day, or 25c of processing!) so I ended up using a combination of Haiku and Sonnet.
Results
For example, here I search PubMed for the "Acarbose" and "longevity", have the LLM read the returned abstracts, and the results look like this:
{
"Summary": "The evidence on whether the compound acarbose has a role in longevity is mixed. There are some promising results from cell-based and animal studies, but the overall evidence is not yet conclusive. More research, especially in human subjects, would be needed to establish a clear role for acarbose in longevity.",
"Compound": "Acarbose",
"Evidence": "Acarbose was identified as one of several longevity-enhancing interventions in a proteomic study of mouse tissues (Burns et al., Geroscience, 2023). Cell-based assays also found that acarbose shared clinically relevant activities with other longevity-enhancing compounds like rapamycin (Venn-Watson et al., Nutrients, 2023). However, the review paper by Elliehausen et al. (BMC Biology, 2023) suggests the relationship between acarbose and exercise for longevity is complex and requires further study.",
"Score": 7
}
To get results formatted as json, Anthropic recommends just asking for json format. In general, all models did a decent job outputting valid json, though Haiku was the worst. Sonnet did a bit better at outputting valid json, and Opus was about the same as Sonnet. As usual, I had to do a bit of semi-manual munging with sed to fix parsing issues.
(As in previous experiments, the first "Summary" entry is a chance for the LLM to think aloud and can be discarded.)
Comparing Haiku and Sonnet
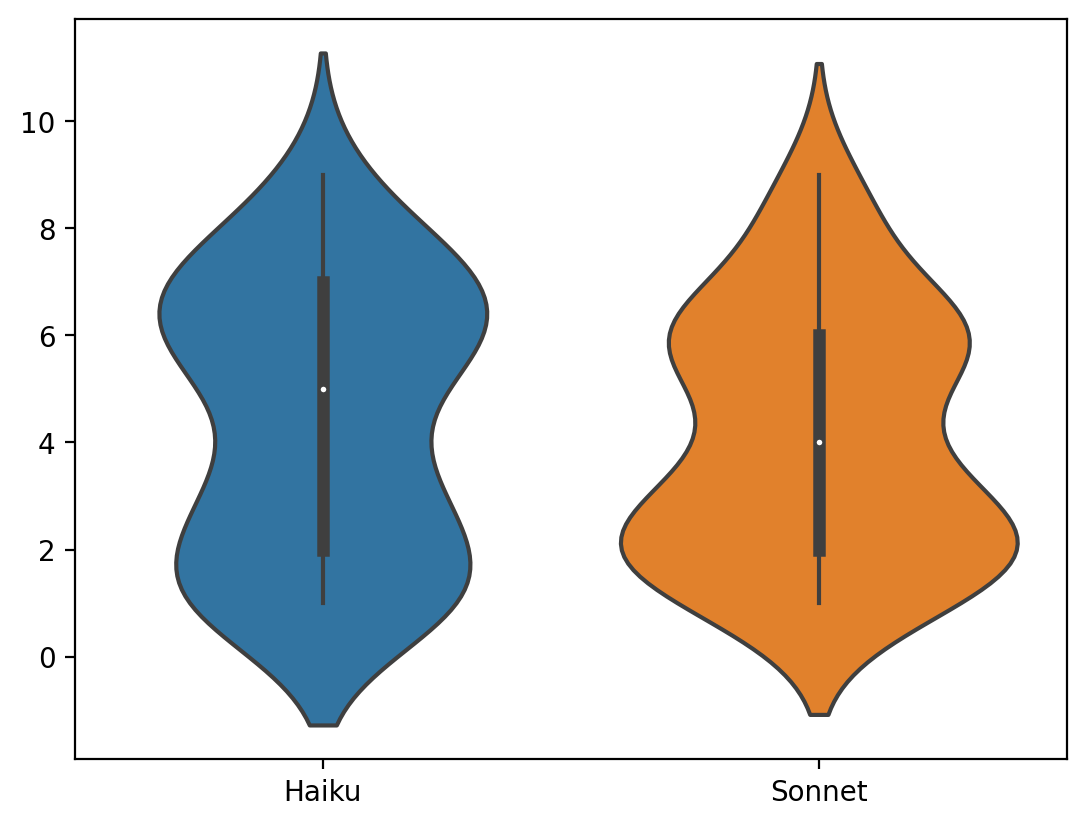

Violin plot and histogram of differences between Sonnet and Haiku for scores > 0
Haiku and Sonnet produced similar scores across the board, with Haiku scores being a little higher. I considered doing a correction to calibrate the scores, or allowing Opus to refine results for the highest scorers, but decided the scores were close enough and there is no gold standard anyway.
Results
I put all 2720 results in a public Google Sheet. 11 compounds score 9/10, 97 score 8/10, 987 score 1-7/10, and 1625 score 0/10 — generally those with no identified literature evidence. Arguably the most interesting stuff will be in the unknown, but also it may make sense to test the drugs with the most evidence as longevity drugs first.
| Compound | Score | Evidence |
|---|---|---|
| Ramipril | 9 | 1) A study in long-lived male mice found that the combination of Ramipril (an ACE inhibitor) and simvastatin (a statin) significantly increased mean and median lifespan by 9%, while the individual therapies were ineffective (Spindler et al., Age, 2016). 2) In a mouse model of Alport syndrome, Ramipril therapy tripled the lifespan when combined with a neutralizing antibody against IL-11, compared to untreated Alport mice (Madison et al., J Pathol, 2024). 3) Another study in Alport mice showed that Ramipril alone can delay the onset of renal failure and reduce renal fibrosis (Gross et al., Kidney Int, 2003). |
| Sarcosine | 9 | Glycine N-methyltransferase (GNMT), which produces sarcosine from glycine, has been shown to extend lifespan in Drosophila by regulating S-adenosylmethionine (SAM) levels (Obata and Miura, Nat Commun, 2015). Sarcosine levels decline with age in rodents and humans, while dietary restriction increases sarcosine levels (Walters et al., Cell Rep, 2018). Sarcosine has been found to induce autophagy, a cellular process associated with longevity (Walters et al., Cell Rep, 2018; Johnson and Cuellar, Ageing Res Rev, 2023). Rilmenidine Phosphate 9 Rilmenidine extends lifespan and healthspan in Caenorhabditis elegans via a nischarin I1-imidazoline receptor (Bennett et al., Aging Cell, 2023) |
| Fulvestrant | 9 | Periodic fasting or a fasting-mimicking diet enhances the activity of the endocrine therapeutic fulvestrant by lowering circulating IGF1, insulin and leptin and inhibiting AKT-mTOR signaling in mouse models of hormone-receptor-positive breast cancer. When combined with palbociclib, adding fasting-mimicking diet cycles promotes long-lasting tumor regression and reverts acquired drug resistance (Caffa et al., Nature, 2020). Betulinic acid 9 Betulinic acid increased lifespan of Drosophila via Sir2 and FoxO activation (Lee and Min, Nutrients, 2024). It mitigated oxidative stress and apoptosis, enhancing longevity in yeast (Sudharshan et al., Free Radic Res, 2022). In C. elegans, it increased lifespan and stress resistance via the insulin/IGF-1 signaling pathway (Chen et al., Front Nutr, 2022). |
| Trametinib (GSK1120212) | 9 | Trametinib, an allosteric inhibitor of MEK in the Ras/MAPK pathway, robustly extends lifespan and reduces age-related gut pathology in Drosophila females by decreasing Pol III activity in intestinal stem cells (Ureña et al., Proc Natl Acad Sci USA, 2024). Trametinib, along with rapamycin and lithium, acts additively to increase longevity in Drosophila by targeting components of the nutrient-sensing network (Castillo-Quan et al., Proc Natl Acad Sci USA, 2019). Trametinib reduces translation errors, and its anti-aging effects are mediated through increased translational fidelity, a mechanism shared with rapamycin and Torin1 (Martinez-Miguel et al., Cell Metab, 2021). The Ras-Erk-ETS signaling pathway is identified as a drug target for longevity, with Trametinib extending lifespan in Drosophila (Slack et al., Cell, 2015). |
| GDC-0941 | 9 | Ortega-Molina et al. (Cell Metab, 2015) showed that pharmacological inhibition of PI3K with GDC-0941 reduced adiposity and metabolic syndrome in obese mice and rhesus monkeys. Bharill et al. (Front Genet, 2013) demonstrated that GDC-0941 extended lifespan and increased stress tolerance in C. elegans, potentially by inhibiting the insulin-like signaling pathway. |
| Hydralazine HCl | 9 | Hydralazine targets cAMP-dependent protein kinase leading to SIRT1/SIRT5 activation and lifespan extension in C. elegans (Dehghan et al., Nature Communications, 2019). Hydralazine induces stress resistance and extends C. elegans lifespan by activating the NRF2/SKN-1 signaling pathway (Dehghan et al., Nature Communications, 2017). Hydralazine might produce anti-aging activity via AMPK activation and help stabilize genomic integrity to prolong life expectancy (Thanapairoje et al., Journal of Basic and Clinical Physiology and Pharmacology, 2023). |
| Geniposide | 9 | 1) Treatment with Gardenia jasminoides Ellis fruit extract (GFE) rich in Geniposide increased the lifespan of C. elegans by 27.1% and improved healthspan markers like pharyngeal pumping, muscle quality, and stress resistance (Choi et al., J Gerontol A Biol Sci Med Sci, 2023). 2) Treatment with 10 mM Geniposide alone increased lifespan by 18.55% and improved healthspan markers in C. elegans (Choi et al., J Gerontol A Biol Sci Med Sci, 2023). 3) Eucommia ulmoides male flower extract (EUFE) containing Geniposide, aucubin, and asperuloside increased C. elegans lifespan by 18.61% and improved healthspan indicators like pharyngeal pumping, mobility, muscle morphology, and stress resistance (Chen et al., Food Funct, 2023). |
| Ivacaftor (VX-770) | 9 | Eur Respir J. (2024) reported that Ivacaftor, in combination with other CFTR modulators, significantly improved lung function (FEV1) and increased the number of adult cystic fibrosis patients, indicating improved longevity. Curr Opin Pulm Med. (2021) highlighted the potential of the triple combination of Ivacaftor, tezacaftor, and elexacaftor to improve lung function, weight, and quality of life in a significant portion of the cystic fibrosis population. J Acad Nutr Diet. (2021) found that Ivacaftor improved weight and BMI in specific CFTR mutation groups. |
| Hesperidin | 9 | Two papers in the Journal of Biomedical Sciences (Yeh et al., 2022; Shen et al., 2024) reported that hesperidin activates the CISD2 gene, which is involved in longevity and healthspan. In naturally aged mice, hesperetin treatment late in life prolonged healthspan and lifespan, ameliorated age-related metabolic decline, and rejuvenated the transcriptome. In human keratinocytes and mouse skin, hesperetin activated CISD2 and attenuated senescence in a CISD2-dependent manner. The 2024 paper in J Biomed Sci also showed that hesperetin can rejuvenate naturally aged skin in mice. |
All results scoring 9/10
There are also some baffling and wrong results. For example, the evidence for Dimethyl Fumarate, which scored 8/10 by Sonnet, clearly points in the wrong direction:
"The paper (Harrison et al., Geroscience, 2024) states: "fisetin, SG1002 (hydrogen sulfide donor), dimethyl fumarate, mycophenolic acid, and 4-phenylbutyrate do not significantly affect lifespan in either sex at the doses and schedules used." It also notes that the amounts of dimethyl fumarate in the diet averaged 35% of the target dose, which may explain the absence of lifespan effects."
Still, overall, the results are good, and the super-cheap Haiku results are indistinguishable from Sonnet. The whole project, even using Sonnet for half, cost less than $10. Some obvious improvements would be widening the scope of the PubMed search beyond search results that directly mention "longevity", or allowing the LLM to read the full text of the papers.
I didn't decide on an intervention to sponsor yet, if any, but I love the crowd-funded aspect of this project, and I hope many more such projects spring up.
Script
The script is a lightly edited variant of the code in a previous blogpost.
import json
import re
import requests
from pathlib import Path
from textwrap import dedent
from time import sleep
from urllib.parse import quote
from tqdm.auto import tqdm
import anthropic
client = anthropic.Anthropic()
# os.environ["ANTHROPIC_API_KEY"] = "sk-xxx"
model, gpt_out_dir = "claude-3-opus-20240229", "out_opus"
model, gpt_out_dir = "claude-3-haiku-20240307", "out_haiku"
model, gpt_out_dir = "claude-3-sonnet-20240229", "out_sonnet"
SEARCH_PUBMED_URL = "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/esearch.fcgi?db=pubmed&term={params}&retmax={max_abstracts}{api_key_param}"
GET_ABSTRACT_URL = "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=pubmed&id={pmid}&retmode=text&rettype=abstract{api_key_param}"
# https://www.ncbi.nlm.nih.gov/research/bionlp/APIs/BioC-PubMed/
BIOC_PMCID_URL = "https://www.ncbi.nlm.nih.gov/research/bionlp/RESTful/pmcoa.cgi/BioC_json/{pmcid}/unicode"
DEFAULT_MAX_ABSTRACTS = 10
# NCBI recommends that users post no more than three URL requests per second and limit large jobs
# Failure to comply with this policy may result in an IP address being blocked from accessing NCBI.
NCBI_API_KEY = None
def remove_ref_blank_entries_and_offsets(obj):
"""
Recursively traverse through the JSON object (dict or list) and:
1. Remove any node if it or any of its nested structures contains a dict with 'section_type': 'REF'.
2. Remove any key-value pairs where the value is an empty list or empty dictionary.
3. Remove any key-value pairs where the key is 'offset'.
"""
if isinstance(obj, dict):
# Check if any value of this dict is a nested dict with 'section_type': 'REF'
if any(isinstance(v, dict) and v.get('section_type') == 'REF' for v in obj.values()):
return None
else:
# No nested dict with 'section_type': 'REF', recursively process each key-value pair
return {k: remove_ref_blank_entries_and_offsets(v) for k, v in obj.items() if k != 'offset' and
remove_ref_blank_entries_and_offsets(v) is not None and v != [] and v != {}}
elif isinstance(obj, list):
# Recursively process each item in the list
return [remove_ref_blank_entries_and_offsets(item) for item in obj
if remove_ref_blank_entries_and_offsets(item) is not None and item != [] and item != {}]
else:
# Return the item as is if it's not a dict or list
return obj
def find_keywords(abstract):
if "longevity" in abstract.lower():
return -10
elif "age" in abstract.lower() or "aging" in abstract.lower():
return -5
else:
return 0
def main(compounds, role, max_abstracts=DEFAULT_MAX_ABSTRACTS, ncbi_api_key=NCBI_API_KEY):
"""Download abstracts and fulltext from pubmed, generate a prompt, query OpenAI.
"""
Path("downloads").mkdir(exist_ok=True)
Path(gpt_out_dir).mkdir(exist_ok=True)
api_key_param = "&api_key={ncbi_api_key}" if ncbi_api_key is not None else ""
for compound in tqdm(compounds, desc="Compounds", leave=True):
abstracts = []
fulltexts = []
params = quote(f'({compound}) AND ("{role}")')
pmtxt = requests.get(SEARCH_PUBMED_URL.format(params=params, max_abstracts=max_abstracts, api_key_param=api_key_param)).text
pmids = re.findall("<Id>(\d+)</Id>", pmtxt)
sleep(0.3)
for pmid in tqdm(pmids, desc="pmids", leave=False):
if Path(f"downloads/abstract.pmid_{pmid}.txt").exists():
abstracts.append(open(f"downloads/abstract.pmid_{pmid}.txt").read())
if Path(f"downloads/fulltext.pmid_{pmid}.json").exists():
fulltexts.append(json.load(open(f"downloads/fulltext.pmid_{pmid}.json")))
continue
abstract = requests.get(GET_ABSTRACT_URL.format(pmid=pmid, api_key_param=api_key_param)).text
open(f"downloads/abstract.pmid_{pmid}.txt", 'w').write(abstract)
abstracts.append(abstract)
sleep(0.3)
pmcid = re.findall("^PMCID:\s+(PMC\S+)$", abstract, re.MULTILINE)
assert len(pmcid) <= 1, "there should be max one PMCID per paper"
pmcid = pmcid[0] if len(pmcid) == 1 else None
if pmcid is not None:
fulltext_request = requests.get(BIOC_PMCID_URL.format(pmcid=pmcid))
if fulltext_request.status_code == 200:
fulltext_json = remove_ref_blank_entries_and_offsets(fulltext_request.json())
json.dump(fulltext_json, open(f"downloads/fulltext.pmid_{pmid}.json", 'w'), indent=2)
fulltexts.append(fulltext_json)
sleep(0.3)
# If there are only a couple of papers then we can include the fulltext
# 250k characters should be well below 100k tokens; 500k characters is too many
abstracts.sort(key=find_keywords)
abstract_len = len("\n".join(abstracts))
fulltext_len = len("\n".join(json.dumps(j) for j in fulltexts))
if abstract_len + fulltext_len < 250_000:
abstracts_txt = "\n".join(abstracts) + "\n" + "\n".join(json.dumps(j) for j in fulltexts)
else:
abstracts_txt = "\n".join(abstracts)
# clip data to the first few abstracts
abstracts_txt = abstracts_txt[:10_000]
gpt_out = f"{gpt_out_dir}/" + f"{compound}_{role}".replace(' ','_').replace('/','_').replace('\\','_') + ".txt"
if len(abstracts_txt) > 1_000 and not Path(gpt_out).exists():
completion = client.messages.create(
model=model,
max_tokens=1024,
system = dedent("""
You are an autoregressive language model that has been fine-tuned with instruction-tuning and RLHF.
You carefully provide accurate, factual, thoughtful, nuanced answers, and are brilliant at reasoning.
If you think there might not be a correct answer, you say so.
Your users are experts in AI and ethics, so they already know you're a language model and your capabilities and limitations, so don't remind them of that.
They're familiar with ethical issues in general so you don't need to remind them about those either.
Don't be verbose in your answers, but do provide details and examples where it might help the explanation.
Your users are also experts in science, and especially biology, medicine, statistics.
Do NOT add any details about how science or research works, tell me to ask my doctor or consult with a health professional.
Do NOT add any details that such an expert would already know.
"""),
messages=[
{"role": "user",
"content": dedent(f"""
Help me research whether this compound has the role of {role}: {compound}.
Here are the top abstracts from pubmed, and any full text that will fit in context:
{abstracts_txt}
Read and synthesize all the evidence from these papers.
Prefer evidence from good journals and highly cited papers, and papers that are recent.
If there is one standout result in a top journal like Nature, Science or Cell, focus on that.
There should usually be one primary result and most of the evidence should depend on that.
Include a score out of 10, where 1 would be a single paper with weak evidence,
5 would be a mid-tier journal with a single paper with believable evidence,
and 10 would be multiple Nature, Science or Cell papers with very strong evidence.
A score above 7 is exceptional, implying at least the result has been replicated and is trustworthy.
Make sure the results are valid json with fields exactly as in the examples below.
Do NOT change the fields, change the order, or add any other fields.
The "Compound" json entry MUST match the query compound. Here, {compound}.
Include AT LEAST ONE reference for each entry on evidence (unless there is no evidence), e.g., (Smith et al., Nature, 2021). ALWAYS include the journal name.
To help you think step-by-step about your response, use the FIRST "Summary" entry to summarize the question
and the relevant available evidence in your own words, in one hundred words or fewer.
Do NOT output markdown, just raw json. The first character should be {{ and the last character should be }}.
Here are some examples of json you might output:
{{
"Summary": "Aspirin has no evidence of {role}. The experiments are not convincing. There is some evidence for a related compound, which may indicate something."; // string
"Compound": "Aspirin", // string
"Evidence": "No evidence as {role}. No relevant papers found.", // string
"Score": 0 // integer from 0-10
}}
{{
"Summary": "There is some evidence for Abacavir, which may indicate something. The evidence is medium, e.g. a paper in a mid-tier journal" // string
"Compound": "Abacavir", // string
"Evidence": "A C. elegans screen identified Abacavir as {role} with a small effect (Jones et al., Nature Cancer, 2001)", // string
"Score": 5 // integer from 0-10
}}
{{
"Summary": "Rapamycin has been cited as {role} many times. The experiments are convincing. The evidence is strong: many papers including a recent Nature paper and a Science paper" // string
"Compound": "Rapamycin" // string
"Evidence": "A cell-based assay identified Rapamycin as {role} in longevity (Smith et al., Science, 2022, Thomson et al., Nature, 2019)", // string
"Score": 10 // integer from 0-10
}}
""")
}
]
)
open(gpt_out, 'w').write(completion.content[0].text)
sleep(.1)
elif not Path(gpt_out).exists():
# just log null results
open(gpt_out, 'w').write("")
if __name__ == "__main__":
compounds = [x.strip() for x in open("compound_list_fixed.txt").readlines()]
main(compounds = compounds, role = "longevity")